Difference between revisions of "Input syntax manual"
(→Input options) |
Ana Jambrina (Talk | contribs) m (→set savesrc) |
||
| Line 1: | Line 1: | ||
| − | + | Serpent has no interactive user interface. All communication between the code and the user is handled through one or several input files and various [[Description of output files|output files]]. | |
| − | + | The format of the input file is unrestricted. The file consists of white-space (space, tab or newline) separated words, containing alphanumeric characters(’a-z’, ’A-Z’, ’0-9’, ’.’, ’-’). If special characters or white spaces need to be used within the word (file names, etc.), the entire string must be enclosed within quotes. | |
| − | + | The input file is divided into separate data blocks, denoted as cards. The file is processed one card at a time and there are no restrictions regarding the order in which the cards should be organized. | |
| + | The input cards are listed [[#input cards|below]]. Additional [[#input options|options]] are followed by key word "set". All input cards and options are case-insensitive (''note to developers: make it so''). Each input card is | ||
| + | delimited by the beginning of the next card. It is hence important that none of the parameter strings used within the card coincide with the card identifiers. | ||
| − | + | The percent-sign ('%') is used to define a comment line. Anything from this character to the end of the line is omitted when the input file is read. Unlike Serpent 1, hashtag ('#') can no longer be used to mark comment lines in Serpent 2 input. The alternative is to use C-style comment sections beginning with "/*" and ending with "*/". Everything between these delimiters is omitted, regardless of the number of newlines or special characters. | |
| − | = | + | This page will contain the whole input syntax of Serpent 2, with links to more detailed descriptions where needed. For reference see also the Serpent 1 input manual<ref name="manual">Leppänen, J. |
| + | ''"Serpent – a Continuous-energy Monte Carlo Reactor Physics Burnup Calculation Code."'' [http://montecarlo.vtt.fi/download/Serpent_manual.pdf User manual], June 18, 2015.</ref>. | ||
| − | === | + | == Input cards<span id="input cards"></span> == |
| − | + | NOTE: Serpent command words are in <tt>'''boldface'''</tt> and input parameters entered by the user in <tt>''CAPITAL ITALIC''</tt>. Optional input parameters are enclosed in [ square brackets ], and when the number of values is not fixed, the remaining values are marked with three dots (...). | |
| − | === | + | === branch (branch definition)<span id="branch"></span> === |
| − | + | '''branch''' ''NAME'' [ '''repm''' ''MAT<sub>1</sub>'' ''MAT<sub>2</sub>'' ] | |
| + | [ '''repu''' ''UNI<sub>1</sub>'' ''UNI<sub>2</sub>'' ] | ||
| + | [ '''stp''' ''MAT DENS TEMP THERM<sub>1</sub> SABL<sub>1</sub> SABH<sub>1</sub> THERM<sub>2</sub> SABL<sub>2</sub> SABH<sub>2</sub> ...'' ] | ||
| + | [ '''tra''' ''TGT TRANS'' ] | ||
| + | [ '''xenon''' ''OPT'' ] | ||
| + | [ '''samarium''' ''OPT'' ] | ||
| + | [ '''norm''' ''NSF'' ] | ||
| + | [ '''gcu''' ''UNI<sub>2</sub>'' ] | ||
| + | [ '''reptrc''' ''FILE<sub>1</sub>'' ''FILE<sub>2</sub>'' ] | ||
| + | [ '''var''' ''VNAME VAL'' ] | ||
| + | Defines the variations invoked for a branch in the automated burnup sequence. Input values: | ||
| − | === src === | + | {| |
| + | | <tt>''NAME''</tt> | ||
| + | | : branch name | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub>''</tt> | ||
| + | | : name of the replaced material | ||
| + | |- | ||
| + | | <tt>''MAT<sub>2</sub>''</tt> | ||
| + | | : name of the replacing material | ||
| + | |- | ||
| + | | <tt>''UNI<sub>1</sub>''</tt> | ||
| + | | : name of the replaced universe | ||
| + | |- | ||
| + | | <tt>''UNI<sub>2</sub>''</tt> | ||
| + | | : name of the replacing universe | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : name of the material for which density and temperature are adjusted | ||
| + | |- | ||
| + | | <tt>''DENS''</tt> | ||
| + | | : material density after adjustment (positive entries for atomic, negative entries for mass densities) | ||
| + | |- | ||
| + | | <tt>''TEMP''</tt> | ||
| + | | : material temperature after adjustment, or -1 if no adjustment in temperature | ||
| + | |- | ||
| + | | <tt>''THERM<sub>n</sub>''</tt> | ||
| + | | : ''n''th thermal scattering data associated with the material | ||
| + | |- | ||
| + | | <tt>''SABL<sub>n</sub>''</tt> | ||
| + | | : name of the ''n''th S(α, β) library for temperature below the given value | ||
| + | |- | ||
| + | | <tt>''SABH<sub>n</sub>''</tt> | ||
| + | | : name of the ''n''th S(α, β) library for temperature above the given value | ||
| + | |- | ||
| + | | <tt>''TGT''</tt> | ||
| + | | : target universe, surface or cell | ||
| + | |- | ||
| + | | <tt>''TRANS''</tt> | ||
| + | | : name of the applied transformation | ||
| + | |- | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option for setting poison concentrations (0 = set to zero, 1 = use values from restart file) | ||
| + | |- | ||
| + | | <tt>''NSF''</tt> | ||
| + | | : normalization scaling factor | ||
| + | |- | ||
| + | | <tt>''FILE<sub>1</sub>''</tt> | ||
| + | | : file path of the replaced transport correction curve data | ||
| + | |- | ||
| + | | <tt>''FILE<sub>2</sub>''</tt> | ||
| + | | : file path of the replacing transport correction curve data | ||
| + | |- | ||
| + | | <tt>''VNAME''</tt> | ||
| + | | : variable name | ||
| + | |- | ||
| + | | <tt>''VAL''</tt> | ||
| + | | : variable value | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The branch name identifies the branch in the coefficient matrix of the [[#coef (coefficient matrix definition)|coef card]] | ||
| + | *The input parameters consist of a number variations, which are invoked when the branch is applied. A single branch card may inclued one or several variations. | ||
| + | *The '''repm''' variation can be used to replace one material with another, for example, to change coolant boron concentration. | ||
| + | **The material replacement works as if <tt>''MAT<sub>1</sub>''</tt> were created using the [[#mat_.28material_definition.29|mat]] or [[#mix_.28mixture_definition.29|mix]] card of <tt>''MAT<sub>2</sub>''</tt>. | ||
| + | **The name of the material present in the geometry will still be <tt>''MAT<sub>1</sub>''</tt> after the replacement, but the material specification (composition, density, tmp, moder, rgb, etc.) will correspond to <tt>''MAT<sub>2</sub>''</tt>. | ||
| + | **This means that all other input-cards that are linked to a specific material name such as [[#det_dm|det dm]], [[#src_sm|src sm]], [[#set_trc|set trc]] and [[#set_iter_nuc|set iter nuc]] can be linked to the original material (<tt>''MAT<sub>1</sub>''</tt>) and they will automatically apply to whatever material <tt>''MAT<sub>2</sub>''</tt> replaces <tt>''MAT<sub>1</sub>''</tt> for the branch calculation. | ||
| + | *The '''repu''' variation can be used to replace one universe with another, for example, to replace empty control rod guide tubes with rodded tubes for control rod insertion in 2D geometries. | ||
| + | **The name of the universe present in the geometry will still be <tt>''UNI<sub>1</sub>''</tt> after the replacement, but the universe contents will correspond to <tt>''UNI<sub>2</sub>''</tt>. | ||
| + | **This means that all other input-cards that are linked to a specific universe name such as [[#det_du|det du]] and [[#src_su|src su]] can be linked to the original universe (<tt>''UNI<sub>1</sub>''</tt>) and they will automatically apply to whatever universe <tt>''UNI<sub>2</sub>''</tt> replaces <tt>''UNI<sub>1</sub>''</tt> for the branch calculation. | ||
| + | *The '''stp''' variation can be used to change material density and temperature. The adjustment is made using the built-in [[Doppler-broadening preprocessor routine]] and tabular interpolation for S(α, β) thermal scattering data. | ||
| + | *The last three parameters of the '''stp''' entry are provided only if the material has thermal scattering libraries attached to it (see the [[#therm (thermal scattering library definition)|therm card]]). | ||
| + | *The '''tra''' variation can be used to move or rotate different parts of the geometry, for example, to adjust the position of control rods in 3D geometries. The name of the transformation refers to the unit (universe, cell or surface) entry in the [[#trans (transformations)|trans card]]. | ||
| + | *The '''xenon''' and '''samarium''' options can be set to enforce the concentrations of fission product poisons Xe-135 and Sm-149 to zero. By default the concentrations are read from the restart file. | ||
| + | *The '''norm''' variation can be used to change the normalization. The adjustment is made applying the parameter ''NSF'' as a multiplicative scaling factor to the given normalization. | ||
| + | *The '''gcu''' variation can be used to replace the universe for group constant generation. This variation is limited to a single-valued GCU-list. | ||
| + | *The '''reptrc''' variation can be used to replace a transport correction file with another. | ||
| + | *Variables can be used to pass information into output file, which may be convenient for the post-processing of the data. | ||
| + | *The branch card is used together with the [[#coef (coefficient matrix definition)|coef card]]. | ||
| + | *For more information, see detailed description on the [[automated burnup sequence]]. | ||
| + | *The replaced material ''MAT<sub>1</sub>'' of '''repm''' variation is also replaced inside mixtures. This means one can not replace a material with a mixture defined with [[#mix (mixture_definition)|mix card]] containing the replaced material (for example replacing pure water defined with [[#mat (material definition)|mat card]] by a mixture of boron and water defined with a [[#mix (mixture definition)|mix card]] containing the same pure water material). | ||
| + | *The replacing material ''MAT<sub>2</sub>'' of '''repm''' variation can not be included in geometry using other cards than the branch card with the '''repm''' variation, version 2.1.30. | ||
| + | *The "sum" option to define the material density as the sum of the constituent nuclide densities is not supported from version 2.2.0 and on. | ||
| + | |||
| + | === casematrix (casematrix definition)<span id="casematrix"></span> === | ||
| + | |||
| + | '''casematrix''' ''CASE_NAME'' | ||
| + | ''NHIS'' [ ''HIS_BR<sub>1</sub>'' ''HIS_BR<sub>2</sub>'' ... ''HIS_BR<sub>NHIS</sub>'' ] | ||
| + | ''NBU'' [ ''BU<sub>1</sub>'' ''BU<sub>2</sub>'' ... ''BU<sub>NBU</sub>'' ] | ||
| + | ''NBR<sub>1</sub>'' [ ''BR<sub>1,1</sub>'' ''BR<sub>1,2</sub>'' ... ''BR<sub>1,NBR<sub>1</sub></sub>'' ] | ||
| + | ''NBR<sub>2</sub>'' [ ''BR<sub>2,1</sub>'' ''BR<sub>2,2</sub>'' ... ''BR<sub>1,NBR<sub>2</sub></sub>'' ] | ||
| + | ... | ||
| + | |||
| + | Defines the casematrix for the automated burnup sequence. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''CASE_NAME''</tt> | ||
| + | | : name of the casematrix | ||
| + | |- | ||
| + | | <tt>''NHIS''</tt> | ||
| + | | : number of history variations | ||
| + | |- | ||
| + | | <tt>''HIS_BR<sub>k</sub>''</tt> | ||
| + | | : name of the ''k''th history variation branch | ||
| + | |- | ||
| + | | <tt>''NBU''</tt> | ||
| + | | : number of burnup points | ||
| + | |- | ||
| + | | <tt>''BU<sub>n</sub>''</tt> | ||
| + | | : burnup steps at which the momentary variation branches are invoked | ||
| + | |- | ||
| + | | <tt>''NBR<sub>m</sub>''</tt> | ||
| + | | : number branches in the ''m''th dimension of the burnup matrix | ||
| + | |- | ||
| + | | <tt>''BR<sub>m,i</sub>''</tt> | ||
| + | | : name of the ''i''th branch in the ''m''th dimension | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The casematrix card performs multiple depletions with <tt>''NHIS''</tt> (different) historical variations and performs restarts similar as the [[#coef|coef]] input card. | ||
| + | *The casematrix card creates a multi-dimensional coefficient matrix (of size <tt>''NBR<sub>1</sub>''</tt> × <tt>''NBR<sub>2</sub>''</tt> × <tt>''NBR<sub>3</sub>''</tt> × ... ). The automated burnup sequence performs a restart for each of the listed burnup points, and loops over the branch combinations defined by the coefficient matrix. This is repeated for each different depletion history. | ||
| + | *Positive values in the burnup vector are interpreted as (MWd/kgU), negative values are interpreted as time steps in days. | ||
| + | *The casematrix card is used together with the [[#branch (branch definition)|branch card]] and [[Installing_and_running_Serpent#Running_casematrix_calculations|-casematrix]] running option. | ||
| + | *Multiple casematrix cards can be given in a single input file. | ||
| + | *For more information, see detailed description on [[automated burnup sequence]]. | ||
| + | |||
| + | === cell (cell definition)<span id="cell"></span> === | ||
| + | |||
| + | |||
| + | '''cell''' ''NAME UNI<sub>0</sub> MAT'' [ ''SURF<sub>1</sub>'' ''SURF<sub>2</sub>'' ... ] | ||
| + | |||
| + | Defines a material cell. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : cell name | ||
| + | |- | ||
| + | | <tt>''UNI<sub>0</sub>''</tt> | ||
| + | | : universe where the cell belongs to | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material that fills the cell | ||
| + | |- | ||
| + | | <tt>''SURF<sub>n</sub>''</tt> | ||
| + | | : surface list | ||
| + | |} | ||
| + | |||
| + | '''cell''' ''NAME UNI<sub>0</sub>'' '''fill''' ''UNI<sub>1</sub>'' [ ''SURF<sub>1</sub>'' ''SURF<sub>2</sub>'' ... ] | ||
| + | |||
| + | Defines a filled cell. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : cell name | ||
| + | |- | ||
| + | | <tt>''UNI<sub>0</sub>''</tt> | ||
| + | | : universe where the cell belongs to | ||
| + | |- | ||
| + | | <tt>''UNI<sub>1</sub>''</tt> | ||
| + | | : universe that fills the cell | ||
| + | |- | ||
| + | | <tt>''SURF<sub>n</sub>''</tt> | ||
| + | | : surface list | ||
| + | |} | ||
| + | |||
| + | '''cell''' ''NAME UNI<sub>0</sub>'' '''outside''' [ ''SURF<sub>1</sub>'' ''SURF<sub>2</sub>'' ... ] | ||
| + | |||
| + | Defines an outside cell. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : cell name | ||
| + | |- | ||
| + | | <tt>''UNI<sub>0</sub>''</tt> | ||
| + | | : universe where the cell belongs to | ||
| + | |- | ||
| + | | <tt>''SURF<sub>n</sub>''</tt> | ||
| + | | : surface list | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *There are three types of cells: material cells, filled cells and outside cells. Filled cells are identified by providing the key word <tt>'''fill'''</tt>, followed by the universe filling the cell. If the key word is missing, the third entry is interpreted as the material filling the cell. Outside cells are identified by replacing the material name with key word <tt>'''outside'''</tt>. | ||
| + | *Cells defined without surfaces are treated as infinite, from version 2.1.32 on. | ||
| + | *Void cells can be defined by setting the material name to "void" | ||
| + | *When the geometry is set up, the root universe must always be defined. By default the root universe is named "0", and it can be changed with the [[#set root|set root]] option. | ||
| + | *Outside cells are used to define the part of the geometry that does not belong to the model. When the particle enters an outside cell, [[#set bc|boundary conditions]] are applied. It is important that the geometry model is non-re-entrant (convex) when vacuum boundary conditions are used. Delta-tracking might miss the boundary conditions in a re-entrant (concave) outer surface. | ||
| + | *Outside cells are allowed only in the root universe. It is important that all space outside the model is defined. | ||
| + | *The surface list defines the boundaries of the cell by listing the surface names (as provided in the [[#surf (surface definition)|surface card]]), together with the operator identifiers (nothing for intersection, ":" for union, "-" for complement and "#" for cell complement). | ||
| + | *Universes are implicitly declared for example by using the <tt>''UNI<sub>0</sub>''</tt> key words on cell cards as there is no explicit universe input card. | ||
| + | *For more information, see detailed description on the [[universe-based geometry type in Serpent]]. | ||
| + | |||
| + | === coef (coefficient matrix definition)<span id="coef"></span> === | ||
| + | |||
| + | '''coef''' ''NBU'' [ ''BU<sub>1</sub>'' ''BU<sub>2</sub>'' ... ] | ||
| + | [ ''NBR<sub>1</sub>'' ''BR<sub>1,1</sub>'' ''BR<sub>1,2</sub>'' ... ] | ||
| + | [ ''NBR<sub>2</sub>'' ''BR<sub>2,1</sub>'' ''BR<sub>2,2</sub>'' ... ] | ||
| + | ... | ||
| + | Defines the coefficient matrix for the automated burnup sequence. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NBU''</tt> | ||
| + | | : number of burnup points | ||
| + | |- | ||
| + | | <tt>''BU<sub>n</sub>''</tt> | ||
| + | | : burnup steps at which the branches are invoked | ||
| + | |- | ||
| + | | <tt>''NBR<sub>m</sub>''</tt> | ||
| + | | : number branches in the ''m''th dimension of the burnup matrix | ||
| + | |- | ||
| + | | <tt>''BR<sub>m,i</sub>''</tt> | ||
| + | | : name of the ''i''th branch in the ''m''th dimension | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The coef card creates a multi-dimensional coefficient matrix (of size <tt>''NBR<sub>1</sub>''</tt> × <tt>''NBR<sub>2</sub>''</tt> × <tt>''NBR<sub>3</sub>''</tt> × ... ). The automated burnup sequence performs a restart for each of the listed burnup points, and loops over the branch combinations defined by the coefficient matrix. | ||
| + | *Positive values in the burnup vector are interpreted as (MWd/kgU), negative values are interpreted as time steps in days. | ||
| + | *The coef card is used together with the [[#branch (branch definition)|branch]] card. | ||
| + | *For multiple historical variations or historical conditions defined using a [[#branch (branch definition)|branch]] card, see the [[#casematrix (casematrix definition)|casematrix]] card. | ||
| + | *For more information, see detailed description on [[automated burnup sequence]]. | ||
| + | |||
| + | === datamesh (general data mesh definition)<span id="datamesh"></span> === | ||
| + | |||
| + | The <tt>'''datamesh'''</tt> card allows the user to define various meshes that can be used for spatial discretization e.g. in [[#det_dmesh|detectors]]. | ||
| + | |||
| + | '''datamesh''' ''NAME'' '''1''' ''USE<sub>LC</sub>'' ''N<sub>X</sub>'' ''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ''N<sub>Y</sub>'' ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ''N<sub>Z</sub>'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' | ||
| + | |||
| + | Defines a regular Cartesian mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of cells in the x direction | ||
| + | |- | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : mesh lower x boundary | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : mesh higher x boundary | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of cells in the y direction | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : mesh lower y boundary | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : mesh higher y boundary | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of cells in the z direction | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : mesh lower z boundary | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : mesh higher z boundary | ||
| + | |} | ||
| + | |||
| + | '''datamesh''' ''NAME'' '''2''' ''USE<sub>LC</sub>'' ''N<sub>R</sub>'' ''R<sub>MIN</sub>'' ''R<sub>MAX</sub>'' ''N<sub>PHI</sub>'' | ||
| + | |||
| + | Defines a regular 2D cylindrical mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''N<sub>R</sub>''</tt> | ||
| + | | : number of cells in the radial direction | ||
| + | |- | ||
| + | | <tt>''R<sub>MIN</sub>''</tt> | ||
| + | | : mesh inner radial boundary | ||
| + | |- | ||
| + | | <tt>''R<sub>MAX</sub>''</tt> | ||
| + | | : mesh outer radial boundary | ||
| + | |- | ||
| + | | <tt>''N<sub>PHI</sub>''</tt> | ||
| + | | : number of cells in the polar angle direction | ||
| + | |} | ||
| + | |||
| + | [[File:Hex lattice x index.png|frame|X-type hexagonal mesh horizontal indexing example for N<sub>X</sub> = N<sub>Y</sub> = 3.]] | ||
| + | '''datamesh''' ''NAME'' '''4''' ''USE<sub>LC</sub>'' ''X<sub>0</sub>'' ''Y<sub>0</sub>'' ''PITCH'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ''N<sub>X</sub>'' ''N<sub>Y</sub>'' ''N<sub>Z</sub>'' | ||
| + | |||
| + | Defines a regular x-type hexagonal mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : mesh horizontal origin x-coordinate | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : mesh horizontal origin y-coordinate | ||
| + | |- | ||
| + | | <tt>''PITCH''</tt> | ||
| + | | : mesh horizontal pitch (equal to cell flat-to-flat width) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : mesh lower z boundary | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : mesh higher z boundary | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of cells in the x direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of cells in the y direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of cells in the z direction | ||
| + | |} | ||
| + | |||
| + | [[File:Hex lattice y index.png|frame|Y-type hexagonal mesh horizontal indexing example for N<sub>X</sub> = N<sub>Y</sub> = 3.]] | ||
| + | '''datamesh''' ''NAME'' '''5''' ''USE<sub>LC</sub>'' ''X<sub>0</sub>'' ''Y<sub>0</sub>'' ''PITCH'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ''N<sub>X</sub>'' ''N<sub>Y</sub>'' ''N<sub>Z</sub>'' | ||
| + | |||
| + | Defines a regular y-type hexagonal mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : mesh horizontal origin x-coordinate | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : mesh horizontal origin y-coordinate | ||
| + | |- | ||
| + | | <tt>''PITCH''</tt> | ||
| + | | : mesh horizontal pitch (equal to cell flat-to-flat width) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : mesh lower z boundary | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : mesh higher z boundary | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of cells in the x direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of cells in the y direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of cells in the z direction | ||
| + | |} | ||
| + | |||
| + | '''datamesh''' ''NAME'' '''6''' ''USE<sub>LC</sub>'' ''N<sub>X</sub>'' ''N<sub>Y</sub>'' ''N<sub>Z</sub>'' ''X<sub>1</sub>'' ... ''X<sub>N<sub>X</sub>+1</sub>'' ''Y<sub>1</sub>'' ... ''Y<sub>N<sub>Y</sub>+1</sub>'' ''Z<sub>1</sub>'' ... ''Z<sub>N<sub>Z</sub>+1</sub>'' | ||
| + | |||
| + | Defines an irregular Cartesian mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of cells in the x direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of cells in the y direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of cells in the z direction | ||
| + | |- | ||
| + | | <tt>''X<sub>i</sub>''</tt> | ||
| + | | : <tt>''N<sub>X</sub>'' + 1</tt> mesh boundaries in the x direction | ||
| + | |- | ||
| + | | <tt>''Y<sub>i</sub>''</tt> | ||
| + | | : <tt>''N<sub>Y</sub>'' + 1</tt> mesh boundaries in the y direction | ||
| + | |- | ||
| + | | <tt>''Z<sub>i</sub>''</tt> | ||
| + | | : <tt>''N<sub>Z</sub>'' + 1</tt> mesh boundaries in the z direction | ||
| + | |} | ||
| + | |||
| + | '''datamesh''' ''NAME'' '''8''' ''USE<sub>LC</sub>'' ''N<sub>R</sub>'' ''N<sub>PHI</sub>'' ''R<sub>1</sub>'' ... ''R<sub>N<sub>R</sub>+1</sub>'' | ||
| + | |||
| + | Defines a radially irregular 2D cylindrical mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''N<sub>R</sub>''</tt> | ||
| + | | : number of cells in the radial direction | ||
| + | |- | ||
| + | | <tt>''N<sub>PHI</sub>''</tt> | ||
| + | | : number of cells in the polar angle direction | ||
| + | |- | ||
| + | | <tt>''R<sub>i</sub>''</tt> | ||
| + | | : <tt>''N<sub>R</sub>'' + 1</tt> mesh boundaries in the r direction | ||
| + | |} | ||
| + | |||
| + | '''datamesh''' ''NAME'' '''9''' ''N<sub>LEVEL</sub>'' ''MESH<sub>1</sub>'' ... ''MESH<sub>N<sub>LEVEL</sub></sub>'' | ||
| + | |||
| + | Defines a regular nested mesh that can be linked to detectors, interfaces etc. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : mesh name | ||
| + | |- | ||
| + | | <tt>''USE<sub>LC</sub>''</tt> | ||
| + | | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search | ||
| + | |- | ||
| + | | <tt>''N<sub>LEVEL</sub>''</tt> | ||
| + | | : number of nested levels in this mesh | ||
| + | |- | ||
| + | | <tt>''MESH<sub>i</sub>''</tt> | ||
| + | | : sub mesh to use at level ''i'' | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *When Serpent makes the mesh search for a specific collision point it will save the collision mesh cell temporarily so that the cell search is conducted at most once even when scoring multiple estimators using the same mesh. | ||
| + | |||
| + | === dep (depletion history)<span id="dep"></span> === | ||
| + | |||
| + | '''dep''' ''STYPE'' [ ''STEP<sub>1</sub> STEP<sub>2</sub> ...'' ] | ||
| + | |||
| + | Defines depletion history with steps and activates depletion calculation mode. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''STYPE''</tt> | ||
| + | | : step type | ||
| + | |- | ||
| + | | <tt>''STEP<sub>n</sub>''</tt> | ||
| + | | : depletion step list | ||
| + | |} | ||
| + | |||
| + | The possible step types are: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Type | ||
| + | ! Description | ||
| + | ! Quantity | ||
| + | ! Unit | ||
| + | |- | ||
| + | | <tt>bustep</tt> | ||
| + | | Depletion step | ||
| + | | Burnup interval | ||
| + | | MWd/kgHM | ||
| + | |- | ||
| + | | <tt>butot</tt> | ||
| + | | Depletion step | ||
| + | | Cumulative burnup | ||
| + | | MWd/kgHM | ||
| + | |- | ||
| + | | <tt>daystep</tt> | ||
| + | | Depletion step | ||
| + | | Time interval | ||
| + | | d | ||
| + | |- | ||
| + | | <tt>daytot</tt> | ||
| + | | Depletion step | ||
| + | | Cumulative time | ||
| + | | d | ||
| + | |- | ||
| + | | <tt>decstep</tt> | ||
| + | | Decay step | ||
| + | | Time interval | ||
| + | | d | ||
| + | |- | ||
| + | | <tt>dectot</tt> | ||
| + | | Decay step | ||
| + | | Cumulative time | ||
| + | | d | ||
| + | |- | ||
| + | | <tt>actstep</tt> | ||
| + | | Activation step | ||
| + | | Time interval | ||
| + | | d | ||
| + | |- | ||
| + | | <tt>acttot</tt> | ||
| + | | Activation step | ||
| + | | Cumulative time | ||
| + | | d | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Transport cycle is omitted with the <tt>decstep</tt> and <tt>dectot</tt> options. | ||
| + | *Transport cycle is run only once with the <tt>actstep</tt> and <tt>acttot</tt> options. | ||
| + | |||
| + | '''dep''' '''pro''' ''REP_NAME'' ''STYPE'' [ ''STEP<sub>1</sub> STEP<sub>2</sub> ...'' ] | ||
| + | |||
| + | Links a reprocessor to the depletion calculation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''REP_NAME''</tt> | ||
| + | | : reprocessor name | ||
| + | |- | ||
| + | | <tt>''STYPE''</tt> | ||
| + | | : step type | ||
| + | |- | ||
| + | | <tt>''STEP<sub>n</sub>''</tt> | ||
| + | | : depletion step list | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The reprocessing system or reprocessor controller is defined using the [[#rep|rep card]]. | ||
| + | |||
| + | '''dep''' '''bra''' ''PTR_BRANCH'' | ||
| + | |||
| + | === det (detector definition)<span id="det"></span> === | ||
| + | |||
| + | '''det''' ''NAME'' [ ''PART'' ] | ||
| + | [ [[#det_dr|'''dr''']] ''MT'' ''MAT'' ] | ||
| + | [ [[#det_dv|'''dv''']] ''VOL'' ] | ||
| + | [ [[#det_dc|'''dc''']] ''CELL'' ] | ||
| + | [ [[#det_du|'''du''']] ''UNI'' ] | ||
| + | [ [[#det_dm|'''dm''']] ''MAT'' ] | ||
| + | [ [[#det_dl|'''dl''']] ''LAT'' ] | ||
| + | [ [[#det_dx|'''dx''']] ''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ''N<sub>X</sub>'' ] | ||
| + | [ [[#det_dy|'''dy''']] ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ''N<sub>Y</sub>'' ] | ||
| + | [ [[#det_dz|'''dz''']] ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ''N<sub>Z</sub>'' ] | ||
| + | [ [[#det_dn|'''dn''']] ''TYPE'' ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''N<sub>1</sub>'' ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ''N<sub>2</sub>'' ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ''N<sub>3</sub>'' ] <small>1/2</small> [ [[#det_dn|'''dn''']] ''TYPE'' ''N<sub>1</sub>'' ''N<sub>2</sub>'' ''N<sub>3</sub>'' ''LIM<sub>11</sub>''...''LIM<sub>1N+1</sub>'' ''LIM<sub>21</sub>''...''LIM<sub>2N+1</sub>'' ''LIM<sub>31</sub>''...''LIM<sub>3N+1</sub>'' ] <small>3/4</small> | ||
| + | [ [[#det_dh|'''dh''']] ''TYPE'' ''X<sub>0</sub>'' ''Y<sub>0</sub>'' ''PITCH'' ''N<sub>1</sub>'' ''N<sub>2</sub>'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ''N<sub>Z</sub>'' ] | ||
| + | [ [[#det_dumsh|'''dumsh''']] ''UNI'' ''N<sub>C</sub>'' ''CELL<sub>0</sub>'' ''BIN<sub>0</sub>'' ''CELL<sub>1</sub>'' ''BIN<sub>1</sub>'' ... ] | ||
| + | [ [[#det_de|'''de''']] ''EGRID'' ] | ||
| + | [ [[#det_di|'''di''']] ''TBIN'' ] | ||
| + | [ [[#det_ds|'''ds''']] ''SURF'' ''DIR'' ] | ||
| + | [ [[#det_dir|'''dir''']] ''COS<sub>X</sub>'' ''COS<sub>Y</sub>'' ''COS<sub>Z</sub>'' ] | ||
| + | [ [[#det_dtl|'''dtl''']] ''SURF'' ] | ||
| + | [ [[#det_df|'''df''']] ''FILE'' ''FRACTION'' ] | ||
| + | [ [[#det_dt|'''dt''']] ''TYPE'' ''PARAM'' ] | ||
| + | [ [[#det_dhis|'''dhis''']] ''OPT'' ] | ||
| + | [ [[#det_dfl|'''dfl''']] ''FLAG'' ''OPT'' ] | ||
| + | [ [[#det_da|'''da''']] ''MAT'' ''FLX'' ] | ||
| + | [ [[#det_dfet|'''dfet''']] ''TYPE'' ''PARAMS'' ] | ||
| + | [ [[#det_dphb|'''dphb''']] ''PHB'' ] | ||
| + | [ [[#det_dmesh|'''dmesh''']] ''MESH'' ] | ||
| + | Detector definition. The first parameter: | ||
| + | |||
| + | {| | ||
| + | | <tt>''PART''</tt> | ||
| + | | : particle type (n = neutron, p = photon) | ||
| + | |} | ||
| + | |||
| + | is optional in single particle simulations. The remaining parameters are defined by separate key words followed by the input values. | ||
| + | |||
| + | |||
| + | Detector response (<tt>'''dr'''</tt>):<span id="det_dr"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MT''</tt> | ||
| + | | : response number | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material name or "<tt>void</tt>" if the material at the collision point is used | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *If the detector is assigned with multiple responses, the results are divided correspondingly into separate bins. | ||
| + | *The response numbers are [[ENDF reaction MT's and macroscopic reaction numbers|ENDF reaction MT's and special reaction types]]. | ||
| + | *Positive response numbers are associated with microscopic cross sections and the result is independent of material density. Materials for microscopic cross sections must consist of a single nuclide. | ||
| + | *Microscopic reactions to ground and isomeric states can be calculated by adding "g" or "m" at the end of the reaction number (e.g. 102g and 102m refer to radiative capture to ground and isomeric states, respectively). This option is available only for nuclides with [[#set_bralib|branching ratios]]. | ||
| + | *Negative response numbers are associated with macroscopic cross sections and special types, and the result is multiplied by material density. | ||
| + | *The response material in the <tt>'''dr'''</tt> entry must not be confused with the material in the <tt>'''dm'''</tt> entry. The former defines the material for the response function, while the latter determines the volume of integration. | ||
| + | *By default, Serpent allows a detector to have at most 10,000,000 bins. | ||
| + | |||
| + | |||
| + | Detector volume (<tt>'''dv'''</tt>):<span id="det_dv"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''VOL''</tt> | ||
| + | | : volume in cm<sup>3</sup> (3D geometry) or cross-sectional area in cm<sup>2</sup> (2D geometry) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The results are divided by detector volume, which is by default set to 1. | ||
| + | |||
| + | |||
| + | Detector cell (<tt>'''dc'''</tt>):<span id="det_dc"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''CELL''</tt> | ||
| + | | : cell name where the detector is scored | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *If multiple detector cells are defined, the results are correspondingly divided into multiple bins. | ||
| + | |||
| + | |||
| + | Detector universe (<tt>'''du'''</tt>):<span id="det_du"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name where the detector is scored | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *If multiple detector universes are defined, the results are correspondingly divided into multiple bins. | ||
| + | |||
| + | |||
| + | Detector material (<tt>'''dm'''</tt>):<span id="det_dm"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material name where the detector is scored | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *If multiple detector materials are defined, the results are correspondingly divided into multiple bins. | ||
| + | *The material entry defines the volume of integration, which must not be confused with the response material in the <tt>'''dr'''</tt> entry. | ||
| + | |||
| + | |||
| + | Detector lattice (<tt>'''dl'''</tt>):<span id="det_dl"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''LAT''</tt> | ||
| + | | : lattice name where the detector is scored | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The lattice detector automatically divides the results into multiple bins corresponding to the lattice cells. | ||
| + | |||
| + | |||
| + | Cartesian mesh detector (<tt>'''dx'''</tt>, <tt>'''dy'''</tt> and <tt>'''dz'''</tt>):<span id="det_dx"></span><span id="det_dy"></span><span id="det_dz"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : minimum x-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : maximum x-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of x-bins | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : minimum y-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : maximum y-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of y-bins | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : minimum z-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : maximum z-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of z-bins | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The mesh detectors can be used to sub-divide the results into multiple spatial bins. For a Cartesian mesh the division is provided with separate entries in x-, y- and z- locations. | ||
| + | |||
| + | |||
| + | Curvilinear and unevenly-spaced mesh detector (<tt>'''dn'''</tt>):<span id="det_dn"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : Type of curvilinear mesh - 1 = cylindrical (dimensions ''r'', ''θ'', ''z''), 2 = spherical (dimensions ''r'', ''θ'', ''φ''), 3 = unevenly-spaced orthogonal (dimensions ''x'', ''y'', ''z''), 4 = unevenly-spaced cylindrical (dimensions ''r'', ''θ'', ''z'') | ||
| + | |- | ||
| + | | <tt>''MIN<sub>n</sub>''</tt> | ||
| + | | : Minimum value of coordinate ''n'' for the mesh division (lengths in cm, angles in degrees). | ||
| + | |- | ||
| + | | <tt>''MAX<sub>n</sub>''</tt> | ||
| + | | : Maximum value of coordinate ''n'' for the mesh division (lengths in cm, angles in degrees). | ||
| + | |- | ||
| + | | <tt>''N<sub>n</sub>''</tt> | ||
| + | | : Number of bins in the ''n'' coordinate direction (the radial division will be equal ''r'', not equal volume, in evenly-spaced types 1/2). | ||
| + | |- | ||
| + | | <tt>''LIM<sub>nm</sub>''</tt> | ||
| + | | : Mesh boundary ''m'' in the ''n'' coordinate direction (lengths in cm, angles in degrees). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *All parameters must be provided, even for one- or two-dimensional curvilinear meshes. | ||
| + | *The results are not divided by cell volume (difference to MCNP mesh tally). | ||
| + | *By default, the curvilinear mesh detectors use the global (universe 0) coordinate system for scoring. If the <tt>''TYPE''</tt> parameter is given as a negative value (e.g. -1) the lowest level coordinates are used instead. | ||
| + | *The syntax for curvilinear (evenly-spaced) mesh detectors (1/-1, 2/-2) differs from the unevenly-spaced mesh ones (3/-3, 4/-4). | ||
| + | |||
| + | |||
| + | Hexagonal mesh detector (<tt>'''dh'''</tt>):<span id="det_dh"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : Type of hexagonal mesh (2 = flat face perpendicular to x-axis, 3 = flat face perpendicular to y-axis) | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt>, <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : coordinates of mesh center | ||
| + | |- | ||
| + | | <tt>''PITCH''</tt> | ||
| + | | : mesh pitch | ||
| + | |- | ||
| + | | <tt>''N<sub>1</sub>''</tt>, <tt>''N<sub>1</sub>''</tt> | ||
| + | | : mesh size | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : minimum z-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : maximum z-coordinate of the detector mesh | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of z-bins | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *All parameters must be provided, even for a two-dimensional hexagonal meshes. | ||
| + | |||
| + | |||
| + | Unstructured mesh detector (<tt>'''dumsh'''</tt>):<span id="det_dumsh"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe of the unstructured mesh based geometry | ||
| + | |- | ||
| + | | <tt>''N<sub>C</sub>''</tt> | ||
| + | | : number of mesh cell bins included in the output | ||
| + | |- | ||
| + | | <tt>''CELL<sub>n</sub>''</tt>, <tt>''BIN<sub>n</sub>''</tt> | ||
| + | | : cell-bin index pairs defining the mapping | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The polyhedral cells in [[Unstructured mesh-based geometry type|unstructured mesh based geometries]] are indexed. | ||
| + | *This detector option allows collecting results from the cells into an arbitrary number of bins. One or multiple cells can be mapped into a single bin. | ||
| + | |||
| + | |||
| + | Detector energy binning (<tt>'''de'''</tt>):<span id="det_de"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''EGRID''</tt> | ||
| + | | : energy grid name | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The results are divided into multiple energy bins based on the grid structure. | ||
| + | *Energy grid structures are defined using the [[#ene (energy grid definition)|ene card]]. [[Pre-defined energy group structures]] can not be directly used in detectors, they have to be redefined using for example the fourth type of ene card. | ||
| + | *The energy boundaries of photon photon pulse-height and photon heat analog detectors are solely defined by the associated energy grid and not limited by the unionized energy grid defining the model. That means that analog detectors might collect scores below the physics model minimum energy bound, without a cut-off, if the energy grid sets it. | ||
| + | |||
| + | |||
| + | Detector time binning (<tt>'''di'''</tt>):<span id="det_di"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''TBIN''</tt> | ||
| + | | : time bin structure name | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The results are divided into multiple time bins. | ||
| + | *Time bin structures are defined using the [[#tme (time bin structure definition)|tme card]]. | ||
| + | *Time bin division may require adjusting the average collision distance ([[#set cfe|set cfe]] option) to achieve sufficient statistical accuracy. | ||
| + | |||
| + | |||
| + | Surface current / flux detector (<tt>'''ds'''</tt>):<span id="det_ds"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''SURF''</tt> | ||
| + | | : surface name | ||
| + | |- | ||
| + | | <tt>''DIR''</tt> | ||
| + | | : direction with respect to surface normal (-2 = flux, -1 = inward current, 1 = outward current, 0 = net current) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *With this option the detector calculates the particle flux over or current through a given surface. | ||
| + | *The surface flux mode is invoked by setting the direction parameter to -2, otherwise this parameter defines the current direction with respect to surface normal. | ||
| + | *Responses are not allowed with current detectors, and with flux detectors, the material name at the collision point has to be specified (<tt>"void"</tt> is not allowed). | ||
| + | *The use of single-bin mesh and cell detectors is allowed to define the integration surface of the detector, from version 2.1.32 on. | ||
| + | *The surface is treated separate from the geometry, and its position is always relative to the origin of the root universe. This is the case even if the surface is part of the geometry in another universe. | ||
| + | *The results are integrated over the surface area (other detectors integrate over volume). | ||
| + | |||
| + | |||
| + | Detector direction vector (<tt>'''dir'''</tt>):<span id="det_dir"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''COS<sub>Y</sub>''</tt> | ||
| + | | : component of the direction vector parallel to x-axis | ||
| + | |- | ||
| + | | <tt>''COS<sub>Y</sub>''</tt> | ||
| + | | : component of the direction vector parallel to y-axis | ||
| + | |- | ||
| + | | <tt>''COS<sub>Z</sub>''</tt> | ||
| + | | : component of the direction vector parallel to z-axis | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option multiplies the detector scores with the scalar product between the particle direction of motion and the given direction vector. | ||
| + | |||
| + | |||
| + | Super-imposed track-length detector (<tt>'''dtl'''</tt>):<span id="det_dtl"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''SURF''</tt> | ||
| + | | : surface inside which the detector is scored | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option can be used to apply the track-length estimator for calculating reaction rates inside regions defined by a single surface (sphere, cylinder, cuboid, etc.) | ||
| + | *The purpose of the track-length detector is to provide better statistics for special applications (activation wire measurements, etc.). | ||
| + | *The surface is treated separate from the geometry, and its position is always relative to the origin of the root universe. This is the case even if the surface is part of the geometry in another universe. | ||
| + | |||
| + | |||
| + | Detector file (<tt>'''df'''</tt>):<span id="det_df"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : file name where the scored points are written | ||
| + | |- | ||
| + | | <tt>''FRAC''</tt> | ||
| + | | : fraction of recorded scores and ascii/binary option (positive value = ascii, negative value = binary) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option can be used to write the scored points in a file. | ||
| + | *When used with the surface current detector this option can provide surface source distributions for other calculations. | ||
| + | *The fraction parameters gives the probability that the score is written in the file and it can be used to reduce the file size in long simulations. | ||
| + | *Source files can be read using the <tt>'''sf'''</tt> entry of [[#src_sf|source cards]]. | ||
| + | |||
| + | |||
| + | Special types (<tt>'''dt'''</tt>):<span id="det_dt"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : special type (see below) | ||
| + | |- | ||
| + | | <tt>''PARAM''</tt> | ||
| + | | : additional parameters | ||
| + | |} | ||
| + | |||
| + | The types are: | ||
| + | {| | ||
| + | | -1 | ||
| + | | = cumulative spectrum | ||
| + | |- | ||
| + | | -2 | ||
| + | | = division by energy width | ||
| + | |- | ||
| + | | -3 | ||
| + | | = division by lethargy width | ||
| + | |- | ||
| + | | -4 | ||
| + | | = sum over cell or material bins | ||
| + | |- | ||
| + | | -5 | ||
| + | | = importance weighting | ||
| + | |- | ||
| + | | -6 | ||
| + | | = sum over number of scores | ||
| + | |- | ||
| + | | 2 | ||
| + | | = multiply result with another detector defined by <tt>''PARAM''</tt> | ||
| + | |- | ||
| + | | 3 | ||
| + | | = divide result with another detector defined by <tt>''PARAM''</tt> | ||
| + | |- | ||
| + | | 4 | ||
| + | | = multiply response function by (local) temperature | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Types -1, -2 and -3 are used with energy binning. | ||
| + | *Type -4 can be used to calculate sum over multiple cell or material bins defined using the <tt>'''dc'''</tt> and <tt>'''dm'''</tt> options. By default separate bins are used for each entry. | ||
| + | *Type 3 can be used to calculate flux-weighted averages (microscopic and macroscopic cross sections, etc.). | ||
| + | *When the results are multiplied or divided by another detector, the number of bins must be compatible (single value or matching number of bins). | ||
| + | |||
| + | |||
| + | History collection option (<tt>'''dhis'''</tt>):<span id="det_dhis"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to collect histories (0 = no, 1 = yes) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *When this option is set, the batch-wise results are printed in the history output file, <tt>[input]_stats.m</tt>. | ||
| + | *''Note to developers: statistical tests should be documented'' | ||
| + | |||
| + | |||
| + | Detector flagging (<tt>'''dfl'''</tt>):<span id="det_dfl"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''FLAG''</tt> | ||
| + | | : flag number (between 1 and 64) | ||
| + | |- | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : flagging option (0 = reset if scored, 1 = set if scored, -2/2 score if set -3/3 score if not set) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Detector flagging allows limiting detector scores to histories which have already contributed to another score. | ||
| + | *The first two options reset or set the flag if the detector is scored, respectively. The remaining options test if the flag is set and score the detector accordingly. Positive values apply OR-type logic (detector is scored if any of the associated flags is set/unset) and negative values AND-type logic (detector is scored if all the associated flags are set/unset). | ||
| + | |||
| + | |||
| + | Activation detector (<tt>'''da'''</tt>):<span id="det_da"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : activated material | ||
| + | |- | ||
| + | | <tt>''FLX''</tt> | ||
| + | | : flux applied to activation | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Activation detector allows performing activation calculation for materials that are not part of the geometry. The flux spectrum applied to neutron irradiation is taken from the detector scores. The absolute flux level can be set using the <tt>''FLX''</tt> parameter. If this parameter is set to -1, also the flux magnitude is taken from the detector scores. | ||
| + | *Requires neutron transport simulation and burnup mode. The material provided with the entry must be burnable, and cannot part of the actual geometry. Volume of the material must be defined using the <tt>'''dv'''</tt> parameter. | ||
| + | *Since the activated material is not part of the physical geometry, this option should be applied only to small samples and other activation calculations in which the isotopic changes do not significantly affect the neutronics. | ||
| + | |||
| + | |||
| + | Functional Expansion Tally detector (<tt>'''dfet'''</tt>):<span id="det_dfet"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : functional expansion type | ||
| + | |- | ||
| + | | <tt>''PARAMS''</tt> | ||
| + | | : other options, specific to each functional expansion type <tt>''TYPE''</tt> | ||
| + | |} | ||
| + | |||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Geometry | ||
| + | ! <tt>PARAMS</tt> | ||
| + | ! <tt>TYPE</tt> | ||
| + | ! Description | ||
| + | ! Functional Series | ||
| + | ! Indexing | ||
| + | |- | ||
| + | | rowspan="1"|Cartesian | ||
| + | | rowspan="1"|<tt>''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ''X<sub>ORDER</sub>'' ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ''Y<sub>ORDER</sub>'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ''Z<sub>ORDER</sub>''</tt> | ||
| + | | <tt>1</tt> | ||
| + | | Legendre only | ||
| + | | <math>\psi(\xi)_n = P_i(\xi_x) P_j(\xi_y) P_k(\xi_z) </math> | ||
| + | | <math>n = k + J * \left( i * I + j\right)</math> | ||
| + | |- | ||
| + | | rowspan="1"|Cylindrical | ||
| + | | rowspan="1"|<tt> ''R<sub>MAX</sub>'' ''R<sub>ORDER</sub>'' ''H<sub>MIN</sub>'' ''H<sub>MAX</sub>'' ''H<sub>ORDER</sub>'' ''H<sub>ORIENTATION</sub>''</tt> | ||
| + | | <tt>2</tt> | ||
| + | | .. | ||
| + | | .. | ||
| + | | .. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *"-1" can be supplied as an <tt>''ORDER''</tt> <tt>PARAM</tt> to use the built-in default values | ||
| + | *It is not recommended to configure a single FET detector to span multiple different material regions—use individual detectors for each region instead | ||
| + | *Specifics of this implementation: | ||
| + | **The FETs are based on nonseparable expansions, i.e. fully-convolved cross terms are included | ||
| + | ***For example, the Legendre-based Cartesian FET uses <math>\psi(\xi)_n = P_i(\xi_x) P_j(\xi_y) P_k(\xi_z) </math> with <math>n</math> as a linear indexer of <math>\{i,j,k\}</math> | ||
| + | ***Due to the properties of orthogonality, these cross terms can be neglected in post-analyses if only separable terms are desired | ||
| + | **A generalization of the Euler formulas for any orthogonal functional series is used | ||
| + | ***The generated FET coefficients <math>a_n</math> already have all contributions from the orthonormalization constant pre-included, i.e. <math>c_n</math> from <math>\frac{1}{c_n} = \lVert \psi_n \rVert^2 = \int_\Gamma \psi_n^2 \omega_n </math> | ||
| + | ***Thus, an FET can be simply reconstructed/sampled from the standard functional series as: <math>F(\xi) = \sum a_n \psi_n(\xi) \omega_n(\xi)</math> | ||
| + | *From version 2.2.0 and on, FET-based detectors follow the standard normalization set in the calculation. The volume standards for detectors are set as default value for FET-based detectors, meaning detectors are not divided by the physical volume (allowing the use of volume detector '''dv'''). | ||
| + | *In version 2.2.0, the relative error evaluation associated with FET-based detectors has been revisited. | ||
| + | |||
| + | Detector pulse-height energy broadening (<tt>'''dphb'''</tt>):<span id="det_dphb"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''PHB''</tt> | ||
| + | | : user-defined (Gaussian) energy broadening for pulse-height detector function name | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * User-defined Gaussian energy broadening functions for pulse height detector are defined using the [[#phb|phb card]]. | ||
| + | |||
| + | |||
| + | Detector spatial integration domain and binning based on a generic data mesh (<tt>'''dmesh'''</tt>):<span id="det_dmesh"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MESH''</tt> | ||
| + | | : name of the [[#datamesh|datamesh]] to use for defining the spatial integration domain and binning for the detector scores | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * Output mesh index will be flattened (one dimensional). | ||
| + | |||
| + | === div (divisor definition)<span id="div"></span> === | ||
| + | |||
| + | '''div''' ''MAT'' [ '''sep''' ''LVL'' ] | ||
| + | [ '''subx''' ''N<sub>X</sub>'' ''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ] <small>equal volume</small> [ '''subr''' ''-N<sub>X</sub>'' ''X<sub>1</sub>'' ''R<sub>2</sub>'' ... ''X<sub>N+1</sub>'' ] <small>manually spaced limits</small> | ||
| + | [ '''suby''' ''N<sub>Y</sub>'' ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ] <small>equal volume</small> [ '''suby''' ''-N<sub>Y</sub>'' ''Y<sub>1</sub>'' ''Y<sub>2</sub>'' ... ''Y<sub>N+1</sub>'' ] <small>manually spaced limits</small> | ||
| + | [ '''subz''' ''N<sub>Z</sub>'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ] <small>equal volume</small> [ '''subz''' ''-N<sub>Z</sub>'' ''Z<sub>1</sub>'' ''Z<sub>2</sub>'' ... ''Z<sub>N+1</sub>'' ] <small>manually spaced limits</small> | ||
| + | [ '''subr''' ''N<sub>R</sub>'' ''R<sub>MIN</sub>'' ''R<sub>MAX</sub>'' ] <small>equal volume</small> [ '''subr''' ''-N<sub>R</sub>'' ''R<sub>1</sub>'' ''R<sub>2</sub>'' ... ''R<sub>N+1</sub>'' ] <small>manually spaced limits</small> | ||
| + | [ '''subs''' ''N<sub>S</sub>'' ''S<sub>0</sub>'' ] <small>equal volume</small> [ '''subs''' ''-N<sub>S</sub>'' ''S<sub>1</sub>'' ''S<sub>2</sub>'' ... ''S<sub>N+1</sub>'' ] <small>manually spaced limits</small> | ||
| + | [ '''lims''' ''FLAG'' ] | ||
| + | Divides a material into a number of sub-zones. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : name of the divided material | ||
| + | |- | ||
| + | | <tt>''LVL''</tt> | ||
| + | | : geometry level at which the cell-wise division takes place (0 = no division, 1 = last level, 2 = 2nd last level, etc.) | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of x-zones | ||
| + | |- | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : minimum x-coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : maximum x-coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''X<sub>n</sub>''</tt> | ||
| + | | : x-coordinate boundaries (cm) | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of y-zones | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : minimum y-coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : maximum y-coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''Y<sub>n</sub>''</tt> | ||
| + | | : y-coordinate boundaries (cm) | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of z-zones | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : minimum z-coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : maximum z-coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''Z<sub>n</sub>''</tt> | ||
| + | | : z-coordinate boundaries (cm) | ||
| + | |- | ||
| + | | <tt>''N<sub>R</sub>''</tt> | ||
| + | | : number of radial zones | ||
| + | |- | ||
| + | | <tt>''R<sub>MIN</sub>''</tt> | ||
| + | | : minimum radial coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''R<sub>MAX</sub>''</tt> | ||
| + | | : maximum radial coordinate (cm) | ||
| + | |- | ||
| + | | <tt>''R<sub>n</sub>''</tt> | ||
| + | | : radial coordinate boundaries (cm) | ||
| + | |- | ||
| + | | <tt>''N<sub>S</sub>''</tt> | ||
| + | | : number of angular sectors | ||
| + | |- | ||
| + | | <tt>''S<sub>0</sub>''</tt> | ||
| + | | : zero position of angular division (degrees) | ||
| + | |- | ||
| + | | <tt>''S<sub>n</sub>''</tt> | ||
| + | | : angular-sector boundaries (degrees) | ||
| + | |- | ||
| + | | <tt>''FLAG''</tt> | ||
| + | | : flag for mapping regions outside (material) limits to divide material - on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The automated divisor feature can be used to sub-divide burnable materials into depletion zones, but the use is not limited to burnup mode. | ||
| + | *The spatial sub-division is based on either Cartesian or cylindrical mesh. | ||
| + | *Volumes of the divided materials must be set manually (see detailed description on [[Defining material volumes|the definition of material volumes]]). | ||
| + | *Using automated instead of manual depletion zone division saves memory, which may become significant in very large burnup calculation problems (see detailed description on [[memory usage#automated depletion zone division|memory usage]]). | ||
| + | *For more information see detailed description on [[automated depletion zone division]]. | ||
| + | *The usage of <tt>''LVL''</tt> is explained on page [[automated depletion zone division]]. | ||
| + | *The feature of mapping regions outside limits is set by default OFF. | ||
| + | *The manually-spaced angular-sector boundaries <tt>''S<sub>n</sub>''</tt> should cover the full/360 degrees angular space. | ||
| + | *If a material is not divided, all occurrences of it are treated as a single depletion zone. For example, if there are multiple fuel pins with same fuel material type, and no ''div card'' is present, all pins are depleted as a single pin. | ||
| + | |||
| + | === dtrans (detector mesh transformation)<span id="dtrans"></span> === | ||
| + | |||
| + | See [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === ene (energy grid definition)<span id="ene"></span> === | ||
| + | |||
| + | '''ene''' ''NAME'' '''1''' ''E<sub>0</sub>'' ''E<sub>1</sub>'' ... | ||
| + | |||
| + | '''ene''' ''NAME'' '''2''' ''N'' ''E<sub>min</sub>'' ''E<sub>max</sub>'' | ||
| + | |||
| + | '''ene''' ''NAME'' '''3''' ''N'' ''E<sub>min</sub>'' ''E<sub>max</sub>'' | ||
| + | |||
| + | '''ene''' ''NAME'' '''4''' ''GRID'' | ||
| + | |||
| + | Defines an energy grid structure. Input values: | ||
| + | |||
| + | {| | ||
| + | |<tt>''NAME''</tt> | ||
| + | |: energy grid name | ||
| + | |- | ||
| + | |<tt>''E<sub>i</sub>''</tt> | ||
| + | |: bin boundaries (type 1) | ||
| + | |- | ||
| + | |<tt>''N''</tt> | ||
| + | |: number of equi-width bins (types 2 and 3) | ||
| + | |- | ||
| + | |<tt>''E<sub>min</sub>''</tt> | ||
| + | |: minimum energy (types 2 and 3) | ||
| + | |- | ||
| + | |<tt>''E<sub>max</sub>''</tt> | ||
| + | |: maximum energy (types 2 and 3) | ||
| + | |- | ||
| + | |<tt>''GRID''</tt> | ||
| + | |: name of the pre-defined grid (type 4) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The first input parameter gives the type (1 = arbitrarily defined, 2 = equal energy-width bins, 3 = equal lethargy-width bins, 4 = [[pre-defined energy group structures|pre-defined energy group structure]]). | ||
| + | *Energy grid structures are used for several purposes, most commonly with [[#det_de|detectors]]. | ||
| + | *See separate description of [[pre-defined energy group structures]]. | ||
| + | |||
| + | === ftrans (fill transformation)<span id="ftrans"></span> === | ||
| + | |||
| + | See [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === fun (function definition)<span id="fun"></span> === | ||
| + | |||
| + | '''fun''' ''NAME'' ''TYPE'' [ ... ] | ||
| + | |||
| + | Defines a function that can be used with detector responses. Input values: | ||
| + | |||
| + | {| | ||
| + | |<tt>''NAME''</tt> | ||
| + | |: function name | ||
| + | |- | ||
| + | |<tt>''TYPE''</tt> | ||
| + | |: function type (currently only supported type is 1 = point-wise tabular data) | ||
| + | |} | ||
| + | |||
| + | The syntax for type 1 is: | ||
| + | |||
| + | '''fun''' ''NAME'' '''1''' ''INTT'' ''X<sub>1</sub>'' ''F<sub>1</sub>'' ''X<sub>2</sub>'' ''F<sub>2</sub>'' ... | ||
| + | |||
| + | where: | ||
| + | {| | ||
| + | |<tt>''INTT''</tt> | ||
| + | |: is the interpolation type (1 = histogram, 2 = lin-lin, 3 = lin-log, 4 = log-lin, 5 = log-log) | ||
| + | |- | ||
| + | |<tt>''X<sub>i</sub>'', ''F<sub>i</sub>''</tt> | ||
| + | |: are the tabulated variable-value pairs | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * The defined function is linked to detector response using [[ENDF reaction MT's and macroscopic reaction numbers|response number]] -100 (syntax: dr -100 ''NAME''). | ||
| + | * The defined function currently is only supported as a flux-based function, aka, flux multiplier. | ||
| + | |||
| + | === hisv (history variation matrix definition)<span id="hisv"></span> === | ||
| + | |||
| + | '''hisv''' [ ''BU<sub>1</sub>'' ''NBR<sub>1</sub>'' ''BR<sub>1,1</sub>'' ''BR<sub>1,2</sub>'' ... ''BR<sub>1,NBR<sub>1</sub></sub>'' ] | ||
| + | [ ''BU<sub>2</sub>'' ''NBR<sub>2</sub>'' ''BR<sub>2,1</sub>'' ''BR<sub>2,2</sub>'' ... ''BR<sub>2,NBR<sub>2</sub></sub>'' ] | ||
| + | ... | ||
| + | Defines the history variation matrix for the automated burnup sequence. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''BU<sub>n</sub>''</tt> | ||
| + | | : burnup steps at which the branches are invoked | ||
| + | |- | ||
| + | | <tt>''NBR<sub>n</sub>''</tt> | ||
| + | | : number branches in the ''n''-th burnup step | ||
| + | |- | ||
| + | | <tt>''BR<sub>n,i</sub>''</tt> | ||
| + | | : name of the ''i''-th branch in the ''n''-th burnup step | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The automated burnup sequence defined by the hisv card follows the same principle as the [[#coef|coef]] input card. | ||
| + | *The hisv card performs multiple depletions within a single depletion calculation following the historical variation sequence, performing a restart at each of the listed burnup points, where it applies the variations defined in the listed branches for the given burnup point. | ||
| + | *Positive values in the burnup vector are interpreted as (MWd/kgU), negative values are interpreted as time steps in days. | ||
| + | *The hisv card is used together with the [[#branch|branch]] card. | ||
| + | |||
| + | === ifc (interface file)<span id="ifc"></span> === | ||
| + | |||
| + | '''ifc''' ''FILE'' ['''setinmat''' ''N<sub>MAT</sub>'' ''MAT<sub>1</sub>'' ''MAT<sub>2</sub>'' ... ''MAT<sub>N<sub>MAT</sub></sub>'' ] | ||
| + | ['''setoutmat''' ''N<sub>MAT</sub>'' ''MAT<sub>1</sub>'' ''MAT<sub>2</sub>'' ... ''MAT<sub>N<sub>MAT</sub></sub>'' ] | ||
| + | |||
| + | Links a [[Multi-physics interface|multi-physics interface]] file to be used with the current input. Input values: | ||
| + | |||
| + | {| | ||
| + | |<tt>''FILE''</tt> | ||
| + | |: path to the multi-physics interface file | ||
| + | |} | ||
| + | |||
| + | The optional cards are explained below. | ||
| + | |||
| + | '''<tt>setinmat</tt>''' adds the possibility to link multiple input materials to the same interface, i.e. the same interface gives temperatures and densities (density factors) for multiple materials. | ||
| + | |||
| + | '''<tt>setoutmat</tt>''' adds the possibility to link multiple output materials to the same interface, i.e. the same interface gives temperatures and densities (density factors) for multiple materials. | ||
| + | |||
| + | {| | ||
| + | |<tt>''N<sub>MAT</sub>''</tt> | ||
| + | |: number of materials to link to the interface | ||
| + | |- | ||
| + | |<tt>''MAT<sub>i</sub>''</tt> | ||
| + | |: name of the ''i''th material linked to the interface | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * If multiple materials are linked to the interface using the '''<tt>setinmat</tt>'''/'''<tt>setoutmat</tt>''' option, the densities in the interface file must be given as density factors, i.e. relative to the material card density (values between 0 and 1). | ||
| + | * If the interface is not updated, '''<tt>setinmat</tt>'''/'''<tt>setoutmat</tt>''' options are not eligible. In the case of regular mesh-based, additionally to not updating the interface: a) the input materials cannot be specified using '''<tt>setinmat</tt>''' if power is tallied in pin-type objects; b) the output materials cannot be specified using '''<tt>setoutmat</tt>''' if power is not tallied on the same mesh. | ||
| + | * Option '''<tt>setinmat</tt>''' was '''<tt>setmat</tt>''' in versions before 2.1.32. | ||
| + | * See also [[Coupled multi-physics calculations]]. | ||
| + | |||
| + | === include (read another input file)<span id="include"></span> === | ||
| + | |||
| + | '''include''' ''FILE'' | ||
| + | Reads another input file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : name of the input file | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The include card can be used to simplify the structure of complicated inputs. | ||
| + | *The input parser starts reading and processing the new file from the point where the input card is placed. Processing of the original file continues after the new file is completed. | ||
| + | *The included file must contain complete input cards and options, it cannot be used to read the values of another card. | ||
| + | |||
| + | === lat (regular lattice definition)<span id="lat"></span> === | ||
| + | |||
| + | See also Section 3.6 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | '''lat''' ''UNI TYPE X<sub>0</sub> Y<sub>0</sub> N<sub>X</sub> N<sub>Y</sub> PITCH'' ''UNI<sub>1</sub> UNI<sub>2</sub> ...'' | ||
| + | |||
| + | Defines a finite two-dimensional lattice in xy-plane with square or X- or Y-type hexagonal elements. The lattice is infinite in z-direction. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name of the lattice | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : lattice type | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : x-coordinate of the lattice origin (origin is in the center of the lattice). | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : y-coordinate of the lattice origin (origin is in the center of the lattice). | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of lattice elements in x-direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of lattice elements in y-direction | ||
| + | |- | ||
| + | | <tt>''PITCH''</tt> | ||
| + | | : lattice pitch | ||
| + | |- | ||
| + | | <tt>''UNI<sub>n</sub>''</tt> | ||
| + | | : list of universes filling the lattice positions | ||
| + | |} | ||
| + | |||
| + | Possible lattice types are: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Type | ||
| + | ! Description | ||
| + | |- | ||
| + | | 1 | ||
| + | | Square lattice | ||
| + | |- | ||
| + | | 2 | ||
| + | | X-type hexagonal lattice | ||
| + | |- | ||
| + | | 3 | ||
| + | | Y-type hexagonal lattice | ||
| + | |- | ||
| + | | 14 | ||
| + | | X-type triangular lattice | ||
| + | |} | ||
| + | |||
| + | [[File:Rect lattice index.png|frame|Lattice type 1 indexing example for N<sub>X</sub> = N<sub>Y</sub> = 3.]] | ||
| + | [[File:Hex lattice x index.png|frame|Lattice type 2 indexing example for N<sub>X</sub> = N<sub>Y</sub> = 3.]] | ||
| + | [[File:Hex lattice y index.png|frame|Lattice type 3 indexing example for N<sub>X</sub> = N<sub>Y</sub> = 3.]] | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Number of universes in list of universes must be ''N<sub>X</sub> × N<sub>Y</sub>''. | ||
| + | *For square lattices the x coordinate increases from left to right and the y coordinate increases from top to bottom, so the first ''N<sub>X</sub>'' values in the list of universes create the bottommost (minimum y) row from minimum x to maximum x and the last ''N<sub>X</sub>'' values in the list of universes create the topmost (maximum y) values. Example of the indexing is provided in the attached figure. | ||
| + | *The line breaks usually present in the list of universes are only used to help visualizing the universe order for the user. Serpent ignores them when processing the list of universes. | ||
| + | *The input of X- and Y-type hexagonal lattices is similar to each other, only the directions of the x- and y-axis change. The axis directions can be checked by using the [[#plot (geometry plot definition)|geometry plotter]]. Examples of the indexing are provided in the attached figures. | ||
| + | |||
| + | '''lat''' ''UNI TYPE X<sub>0</sub> Y<sub>0</sub> PITCH UNI<sub>1</sub>'' | ||
| + | |||
| + | Defines an infinite two-dimensional lattice in xy-plane with infinitely repeating square or X- or Y-type hexagonal element. The lattice is infinite in z-direction. | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name of the lattice | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : lattice type | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : x-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : y-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''PITCH''</tt> | ||
| + | | : lattice pitch | ||
| + | |- | ||
| + | | <tt>''UNI<sub>1</sub>''</tt> | ||
| + | | : universe name of the universe filling all lattice positions | ||
| + | |} | ||
| + | |||
| + | Possible lattice types are: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Type | ||
| + | ! Description | ||
| + | |- | ||
| + | | 6 | ||
| + | | Square lattice | ||
| + | |- | ||
| + | | 7 | ||
| + | | Y-type hexagonal lattice | ||
| + | |- | ||
| + | | 8 | ||
| + | | X-type hexagonal lattice | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The order of X- and Y-type hexagonal lattice type numbers is reversed when compared with finite hexagonal lattices. | ||
| + | |||
| + | '''lat''' ''UNI TYPE X<sub>0</sub> Y<sub>0</sub> N<sub>R</sub>'' ''N<sub>S,1</sub> RADIUS<sub>1</sub> THETA<sub>1</sub> UNI<sub>1,1</sub> UNI<sub>2,1</sub> ... N<sub>S,2</sub> RADIUS<sub>2</sub> THETA<sub>2</sub> UNI<sub>1,2</sub> UNI<sub>2,2</sub> ... ...'' | ||
| + | |||
| + | Defines a finite two-dimensional circular cluster array lattice in xy-plane. The lattice is infinite in z-direction. | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name of the lattice | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : lattice type | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : x-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : y-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''N<sub>R</sub>''</tt> | ||
| + | | : number of rings in the array | ||
| + | |- | ||
| + | | <tt>''N<sub>S,R</sub>''</tt> | ||
| + | | : number of sectors in Rth ring | ||
| + | |- | ||
| + | | <tt>''RADIUS<sub>R</sub>''</tt> | ||
| + | | : central radius of Rth ring | ||
| + | |- | ||
| + | | <tt>''THETA<sub>R</sub>''</tt> | ||
| + | | : angle of rotation of Rth ring in degrees | ||
| + | |- | ||
| + | | <tt>''UNI<sub>N,R</sub>''</tt> | ||
| + | | : list of universes filling the sector positions in Rth ring | ||
| + | |} | ||
| + | |||
| + | Possible lattice type is: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Type | ||
| + | ! Description | ||
| + | |- | ||
| + | | 4 | ||
| + | | Circular cluster array | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The circular cluster array can be used to define fuel assemblies used for example in AGR, CANDU, MAGNOX and RBMK reactors. It can also be used to define fuel rod layout used for example in TRIGA reactors. | ||
| + | |||
| + | '''lat''' ''UNI TYPE X<sub>0</sub> Y<sub>0</sub> N<sub>L</sub> Z<sub>1</sub> UNI<sub>1</sub> Z<sub>2</sub> UNI<sub>2</sub> ...'' | ||
| + | |||
| + | Defines a finite one-dimensional vertical stack in z-direction. The stack is infinite in xy-plane. | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name of the lattice | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : lattice type | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : x-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : y-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''N<sub>L</sub>''</tt> | ||
| + | | : number of lattice elements in z-direction (number of axial layers) | ||
| + | |- | ||
| + | | <tt>''Z<sub>n</sub>''</tt> | ||
| + | | : z-coordinate of the nth lattice element (lower boundary of the axial layer) | ||
| + | |- | ||
| + | | <tt>''UNI<sub>n</sub>''</tt> | ||
| + | | : universe name filling the nth lattice position | ||
| + | |} | ||
| + | |||
| + | Possible lattice type is: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Type | ||
| + | ! Description | ||
| + | |- | ||
| + | | 9 | ||
| + | | Vertical stack | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The z-coordinates must be given in ascending order. | ||
| + | *Space below the lowest z-coordinate is not defined. | ||
| + | *The top layer fills the entire space above the highest z-coordinate. | ||
| + | *The number of ''Z<sub>n</sub>-UNI<sub>n</sub>'' pairs must be ''N<sub>L</sub>''. | ||
| + | |||
| + | '''lat''' ''UNI TYPE X<sub>0</sub> Y<sub>0</sub> Z<sub>0</sub> N<sub>X</sub> N<sub>Y</sub> N<sub>Z</sub> PITCH<sub>X</sub> PITCH<sub>Y</sub> PITCH<sub>Z</sub> UNI<sub>1</sub> UNI<sub>2</sub> ...'' | ||
| + | |||
| + | Defines a finite three-dimensional lattice in xyz-space with cuboidal or X- or Y-type hexagonal prism elements. | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name of the lattice | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : lattice type | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : x-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : y-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''Z<sub>0</sub>''</tt> | ||
| + | | : z-coordinate of the lattice origin | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of lattice elements in x-direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of lattice elements in y-direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of lattice elements in z-direction | ||
| + | |- | ||
| + | | <tt>''PITCH<sub>X</sub>''</tt> | ||
| + | | : lattice pitch in x-direction | ||
| + | |- | ||
| + | | <tt>''PITCH<sub>Y</sub>''</tt> | ||
| + | | : lattice pitch in y-direction | ||
| + | |- | ||
| + | | <tt>''PITCH<sub>Z</sub>''</tt> | ||
| + | | : lattice pitch in z-direction | ||
| + | |- | ||
| + | | <tt>''UNI<sub>n</sub>''</tt> | ||
| + | | : list of universes filling the lattice positions | ||
| + | |} | ||
| + | |||
| + | Possible lattice types are: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Type | ||
| + | ! Description | ||
| + | |- | ||
| + | | 11 | ||
| + | | Cuboidal lattice | ||
| + | |- | ||
| + | | 12 | ||
| + | | X-type hexagonal prism lattice | ||
| + | |- | ||
| + | | 13 | ||
| + | | Y-type hexagonal prism lattice | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Number of universes in list of universes must be ''N<sub>X</sub> × N<sub>Y</sub> × N<sub>Z</sub>''. | ||
| + | *For hexagonal prism lattices the x- and y-direction pitches must be equal. | ||
| + | *The universe indexing is the similar as with lattice types 1-3. The lowermost z-level is given first, and the uppermost z-level is given last. | ||
| + | |||
| + | === ltrans (lattice transformation)<span id="ltrans"></span> === | ||
| + | |||
| + | See [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === mat (material definition)<span id="mat"></span> === | ||
| + | |||
| + | See Chapter 4 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | '''mat''' ''NAME DENS'' [ [[#mat_tmp|'''tmp''']] ''TEMP'' ] | ||
| + | [ [[#mat_tms|'''tms''']] ''TEMP'' ] | ||
| + | [ [[#mat_tft|'''tft''']] ''T<sub>MIN</sub>'' ''T<sub>MAX</sub>'' ] | ||
| + | [ [[#mat_rgb|'''rgb''']] ''R G B'' ] | ||
| + | [ [[#mat_vol|'''vol''']] ''VOL'' ] | ||
| + | [ [[#mat_mass|'''mass''']] ''MASS'' ] | ||
| + | [ [[#mat_burn|'''burn''']] ''N<sub>R</sub>'' ] | ||
| + | [ [[#mat_fix|'''fix''']] ''ID'' ''TEMP'' ] | ||
| + | [ [[#mat_moder|'''moder''']] ''THNAME'' ''ZA'' ] | ||
| + | ''NUC<sub>1</sub> FRAC<sub>1</sub>'' | ||
| + | [ ''NUC<sub>2</sub> FRAC<sub>2</sub>'' ] | ||
| + | [ ''...'' ] | ||
| + | |||
| + | <u>Mandatory information:</u> | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : Name of the material | ||
| + | |- | ||
| + | | <tt>''DENS''</tt> | ||
| + | | : Density of the material (positive for atomic, negative for mass density) or '''<tt>sum</tt>''' to calculate the density from given nuclide fractions | ||
| + | |- | ||
| + | | <tt>''NUC<sub>n</sub>''</tt> | ||
| + | | : Identifier of ''n''th nuclide in composition, e.g. "92235.03c" or "U-235.03c". | ||
| + | |- | ||
| + | | <tt>''FRAC<sub>n</sub>''</tt> | ||
| + | | : Fraction of ''n''th nuclide in composition, positive values are interpreted as atomic fractions/densities, negative values as mass fractions/densities. | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Optional cards:</u> | ||
| + | |||
| + | '''tmp''': <span id="mat_tmp"></span> Material temperature for [[Doppler-broadening preprocessor routine|Doppler-preprocessor]] | ||
| + | |||
| + | {| | ||
| + | | <tt>''TEMP''</tt> | ||
| + | | : Temperature (in Kelvin) of the material for [[Doppler-broadening preprocessor routine|Doppler-broadening preprocessor]] | ||
| + | |} | ||
| + | |||
| + | '''tms''': <span id="mat_tmp"></span> Material temperature for on-the-fly [[TMS on-the-fly temperature treatment routine|TMS temperature treatment]] | ||
| + | |||
| + | {| | ||
| + | | <tt>''TEMP''</tt> | ||
| + | | : Temperature (in Kelvin) of the material for on-the-fly [[TMS on-the-fly temperature treatment routine|TMS temperature treatment]] | ||
| + | |} | ||
| + | |||
| + | '''tft''': <span id="mat_tft"></span> Temperature limits for material for [[Coupled multi-physics calculations|coupled multi-physics calculations]] | ||
| + | |||
| + | {| | ||
| + | | <tt>''T<sub>MIN</sub>''</tt> | ||
| + | | : Lower limit for material temperature | ||
| + | |- | ||
| + | | <tt>''T<sub>MAX</sub>''</tt> | ||
| + | | : Upper limit for material temperature | ||
| + | |} | ||
| + | |||
| + | '''rgb''': <span id="mat_rgb"></span> Material color for [[#plot|geometry plots]] | ||
| + | |||
| + | {| | ||
| + | | <tt>''R''</tt> | ||
| + | | : Value for the red channel of [[#plot|geometry plots]] (between 0 and 255) | ||
| + | |- | ||
| + | | <tt>''G''</tt> | ||
| + | | : Value for the green channel of geometry plots (between 0 and 255) | ||
| + | |- | ||
| + | | <tt>''B''</tt> | ||
| + | | : Value for the blue channel of geometry plots (between 0 and 255) | ||
| + | |} | ||
| + | |||
| + | '''vol''': <span id="mat_vol"></span> Material volume | ||
| + | |||
| + | {| | ||
| + | | <tt>''VOL''</tt> | ||
| + | | : Volume of the material in cm<sup>3</sup> (3D geometry) or cross-sectional area in cm<sup>2</sup> (2D geometry) | ||
| + | |} | ||
| + | |||
| + | '''mass''': <span id="mat_mass"></span> Material mass | ||
| + | |||
| + | {| | ||
| + | | <tt>''MASS''</tt> | ||
| + | | : Mass of the material in grams | ||
| + | |} | ||
| + | |||
| + | '''burn''': <span id="mat_burn"></span> Flag material for depletion | ||
| + | |||
| + | {| | ||
| + | | <tt>''N<sub>R</sub>''</tt> | ||
| + | | : Set to 1 in order to deplete material. The depletion zone division should be done using the [[#div|div]]-card. | ||
| + | |} | ||
| + | |||
| + | '''fix''': <span id="mat_fix"></span> Library information for decay nuclides | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB''</tt> | ||
| + | | : Library ID (e.g. "09c") for nuclides without cross section data. | ||
| + | |- | ||
| + | | <tt>''TEMP''</tt> | ||
| + | | : Temperature (in Kelvin) for nuclides without cross section data. | ||
| + | |} | ||
| + | |||
| + | '''moder''': <span id="mat_moder"></span> Use thermal scattering data library for a nuclide. The moder entry can be used several times to define thermal scattering libraries for multiple nuclides, such as hydrogen and deuterium in heavy water. | ||
| + | |||
| + | {| | ||
| + | | <tt>''THNAME''</tt> | ||
| + | | : Name of the thermal scattering data library defined using the [[#therm_and_thermstoch_.28thermal_scattering.29|therm]] card. | ||
| + | |- | ||
| + | | <tt>''ZA''</tt> | ||
| + | | : Nuclide ZA of the thermal scatterer (e.g. 1001 for H-1). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This description is incomplete for both the descriptions and optional settings. | ||
| + | *See [[defining material volumes]] and [[#set mvol|set mvol]] regarding other ways to set the material volumes for example in burnup calculations. | ||
| + | *The nuclide identifier for nuclides with associated cross-sections corresponds to ZZAAA.ID and, for nuclides without associated cross-sections, e.g., decay nuclides, to ZZAAAI. The identifiers include ''Z'', the atomic number; ''A'', the mass number of the nuclide; ''I'', the isomeric state (0 = ground state, 1 = metastable state); and ''ID'', the library identifier. For nuclides without associated cross-sections, include the '''fix''' option to indicate the library and temperature of the given nuclides. | ||
| + | |||
| + | === mesh (mesh plot definition)<span id="mesh"></span> === | ||
| + | |||
| + | '''mesh''' ''ORI'' ''XPIX'' ''YPIX'' [ ''SYM'' ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ] | ||
| + | |||
| + | '''mesh''' 8 ''CMAP'' ''DET'' ''ORI'' ''XPIX'' ''YPIX'' [ ''SYM'' ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ] | ||
| + | |||
| + | '''mesh''' 10 ''ORI'' ''XPIX'' ''YPIX'' | ||
| + | |||
| + | Produces a png-format mesh plot of various results. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''ORI''</tt> | ||
| + | | : orientation with respect to coordinate axes | ||
| + | |- | ||
| + | | <tt>''XPIX''</tt> | ||
| + | | : horizontal image size in pixels | ||
| + | |- | ||
| + | | <tt>''YPIX''</tt> | ||
| + | | : vertical image size in pixels | ||
| + | |- | ||
| + | | <tt>''SYM''</tt> | ||
| + | | : symmetry option (not used in Serpent 2) | ||
| + | |- | ||
| + | | <tt>''MIN<sub>n</sub>'' ''MAX<sub>n</sub>''</tt> | ||
| + | | : boundaries of the plotted region | ||
| + | |- | ||
| + | | <tt>''CMAP''</tt> | ||
| + | | : color map used for plotting detector scores (positive entry for linear, negative for log-scale) | ||
| + | |- | ||
| + | | <tt>''DET''</tt> | ||
| + | | : detector name | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The first format produces a mesh plot where fission rate and thermal flux distribution are plotted using hot and cold color schemes, respectively. This type of mesh plot is convenient for illustrating the neutronics of thermal systems. The orientation parameter defines the coordinate axis perpendicular to the plot plane: 1 - x-axis (projection on yz-plane); 2 - y-axis (projection on xz-plane); 3 - z-axis (projection on xy-plane). | ||
| + | *The second format can be used for plotting scores that contribute to a detector. The additional input parameters are the detector name and the color map used in the plot. | ||
| + | *The third format generates a mesh-plot of the temperature distribution. This can be a good way to check the temperature distribution, provided by an external solver, during a coupled calculation. | ||
| + | *The color maps are: 1 - hot; 2 - cold; 4 - jet; 5 - black and white; 6 - hsv; 7 - spring; 8 - summer; 9 - autumn; 10 - winter; 11 - green-purple; 12 - purple-orange; 13 - blue-red. Many of these correspond to what is used in Matlab. Logarithmic scale is used if the number is given with a minus sign. | ||
| + | *Detector scores are collected in the mesh cells (see the [[#det (detector definition)|detector card]] and the [[ENDF reaction MT's and macroscopic reaction numbers|list of ENDF reaction MT's and special reaction types]] for more information). The distribution is scaled according to the minimum and maximum values. | ||
| + | *Some special detector types, such as pulse-height detectors and analog photon heating detectors cannot be associated with mesh plots. | ||
| + | *The mesh plot always produces results that are integrated over space. If no boundaries are provided, the integration is carried over the entire geometry. | ||
| + | *Setting the orientation parameter of a detector mesh plot to 4 produces a plot in cylindrical coordinates. Instead of Cartesian boundaries the entered values are then the radius, angle and axial coordinate. | ||
| + | *The symmetry option was used in Serpent 1. The parameter must be provided for Serpent 2 as well, even though it is not used. The value can be set to zero. | ||
| + | *Mesh plot produced by the nth mesh-card is written in file <tt>[input]_mesh[n].png</tt>. | ||
| + | |||
| + | === mflow (material flow definition)<span id="mflow"></span> === | ||
| + | |||
| + | '''mflow''' ''NAME'' | ||
| + | ''NUC<sub>1</sub>'' ''λ<sub>1</sub>'' | ||
| + | [ ''NUC<sub>2</sub>'' ''λ<sub>2</sub>'' ] | ||
| + | [ ''...'' ] | ||
| + | |||
| + | Defines the material flow. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : name of the material flow | ||
| + | |- | ||
| + | | <tt>''NUC<sub>n</sub>''</tt> | ||
| + | | : identifier of <tt>''n''</tt>th nuclide in composition | ||
| + | |- | ||
| + | | <tt>''λ''<sub>n</sub></tt> | ||
| + | | : reprocessing constant of <tt>''n''</tt>th nuclide in composition (in s<sup>-1</sup>) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | * The nuclide ID can be replaced with "all", in which case all nuclides are included with the same reprocessing fraction ''λ''. | ||
| + | * The nuclide ID should follow the ZAI or ISO format (e.g., 922350 or U-235). | ||
| + | |||
| + | === mix (mixture definition)<span id="mix"></span> === | ||
| + | |||
| + | '''mix''' ''NAME'' [ '''rgb''' ''R G B'' ] | ||
| + | [ '''vol''' ''VOL'' ] | ||
| + | [ '''mass''' ''MASS'' ] | ||
| + | ''MAT<sub>1</sub>'' ''F<sub>1</sub>'' | ||
| + | ''MAT<sub>2</sub>'' ''F<sub>2</sub>'' | ||
| + | ... | ||
| + | Defines a mixture of two or several materials. Input values: | ||
| + | |||
| + | <u>Mandatory information:</u> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>n</sub>''</tt> | ||
| + | | : material name | ||
| + | |- | ||
| + | | <tt>''F<sub>n</sub>''</tt> | ||
| + | | : material fraction (positive values for volume, negative values for mass fractions) | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Optional cards:</u> | ||
| + | |||
| + | '''rgb''': <span id="mat_rgb"></span> Material color for [[#plot|geometry plots]] | ||
| + | |||
| + | {| | ||
| + | | <tt>''R''</tt> | ||
| + | | : Value for the red channel of [[#plot|geometry plots]] (between 0 and 255) | ||
| + | |- | ||
| + | | <tt>''G''</tt> | ||
| + | | : Value for the green channel of geometry plots (between 0 and 255) | ||
| + | |- | ||
| + | | <tt>''B''</tt> | ||
| + | | : Value for the blue channel of geometry plots (between 0 and 255) | ||
| + | |} | ||
| + | |||
| + | '''vol''': <span id="mat_vol"></span> Material volume | ||
| + | |||
| + | {| | ||
| + | | <tt>''VOL''</tt> | ||
| + | | : Volume of the material in cm<sup>3</sup> (3D geometry) or cross-sectional area in cm<sup>2</sup> (2D geometry) | ||
| + | |} | ||
| + | |||
| + | '''mass''': <span id="mat_mass"></span> Material mass | ||
| + | |||
| + | {| | ||
| + | | <tt>''MASS''</tt> | ||
| + | | : Mass of the material in g | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Mixtures can be used to define complicated material definitions consisting of two or more physical materials mixed homogeneously. | ||
| + | *Serpent decomposes these mixtures into standard materials before running the transport simulation. | ||
| + | *The decomposed material compositions can be written into file using the [[Installing and running Serpent#Running Serpent|-mix]] command line option. | ||
| + | *Nuclide specific [[#mat_moder|thermal scattering data]] is automatically brought from component materials to the mixture. | ||
| + | *Many other input cards such as [[#set_trc|set trc]], [[#set_iter_nuc|set iter nuc]], [[Sensitivity_calculations#Choosing_materials_to_perturb|sens pert matlist]] are not automatically inherited by the mixture from the components and should be directly defined using the mixture material name (opposed to component material names) if they are to be applied to the mixture. | ||
| + | |||
| + | === nest (nested universe definition)<span id="nest"></span> === | ||
| + | |||
| + | '''nest''' ''U'' ''TYPE'' | ||
| + | [ ''MAT<sub>1</sub>'' ''R<sub>1</sub>'' ] | ||
| + | [ ''MAT<sub>2</sub>'' ''R<sub>2</sub>'' ] | ||
| + | ... | ||
| + | [ ''MAT<sub>N</sub>'' ] | ||
| + | |||
| + | '''nest''' ''U'' | ||
| + | [ ''MAT<sub>1</sub>'' ''TYPE<sub>1</sub>'' ''PARAM<sub>11</sub> PARAM<sub>12</sub>'' ... ] | ||
| + | [ ''MAT<sub>2</sub>'' ''TYPE<sub>2</sub>'' ''PARAM<sub>21</sub> PARAM<sub>22</sub>'' ... ] | ||
| + | ... | ||
| + | [ ''MAT<sub>N</sub>'' ] | ||
| + | |||
| + | Defines a universe consisting of nested regions. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''U''</tt> | ||
| + | | : universe name | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : nested surface type (single surface for all regions) | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub> ... MAT<sub>N</sub>''</tt> | ||
| + | | : material regions | ||
| + | |- | ||
| + | | <tt>''R<sub>1</sub> ... R<sub>N-1</sub>''</tt> | ||
| + | | : outer radii | ||
| + | |- | ||
| + | | <tt>''TYPE<sub>1</sub> ... TYPE<sub>N-1</sub>''</tt> | ||
| + | | : nested surface type (different surfaces for each region) | ||
| + | |- | ||
| + | | <tt>''PARAM<sub>nm</sub> ... </tt> | ||
| + | | : surface parameters | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The nest card defines an entire universe consisting of nested material regions. The boundaries are defined by surfaces nested inside each other. The outermost region is infinite. | ||
| + | *The material entries can be replaced by <tt>'''fill''' ''U<sub>0</sub>''</tt>, in which case the region is filled by another universe. | ||
| + | *The first format allows defining nests in which all surfaces are of same type and centred at the origin. Only surfaces that are characterized by a single outer radius are accepted ([[Surface_types#Second-order_quadratic_surfaces|cylinders, spheres]] and some [[Surface_types#Regular_prisms|regular prisms]]). The [[#pin (pin geometry definition)|pin]] and [[#particle (particle geometry definition)|particle]] definitions are short-hand notations of the nest card. | ||
| + | *The second format allows mixing different surface types. In this case all surface parameters need to be provided after the surface type. | ||
| + | |||
| + | === particle (particle geometry definition)<span id="particle"></span> === | ||
| + | |||
| + | '''particle''' ''U'' | ||
| + | [ ''MAT<sub>1</sub>'' ''R<sub>1</sub>'' ] | ||
| + | [ ''MAT<sub>2</sub>'' ''R<sub>2</sub>'' ] | ||
| + | ... | ||
| + | [ ''MAT<sub>N</sub>'' ] | ||
| + | Defines a particle universe. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''U''</tt> | ||
| + | | : universe name | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub> ... MAT<sub>N</sub>''</tt> | ||
| + | | : material regions | ||
| + | |- | ||
| + | | <tt>''R<sub>1</sub> ... R<sub>N-1</sub>''</tt> | ||
| + | | : outer radii | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The particle card defines an entire universe consisting of nested spherical shells. The boundaries are defined by sphere surfaces. The outermost region is radially infinite. | ||
| + | *The material entries can be replaced by <tt>'''fill''' ''U<sub>0</sub>''</tt>, in which case the region is filled by another universe. | ||
| + | *Most typically used for defining TRISO fuel particles. | ||
| + | *The particle card is special case of a [[#nest (nested universe definition)|nested universe type]]. | ||
| + | *See also description of [[#pbed (explicit stochastic geometry)|explicit stochastic geometry type]]. | ||
| + | |||
| + | === pbed (explicit stochastic (pebble bed) geometry) === | ||
| + | |||
| + | '''pbed''' ''U<sub>0</sub>'' ''U<sub>bg</sub>'' ''FILE'' [ ''OPT'' ] | ||
| + | Defines a stochastic particle / pebble-bed geometry. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''U<sub>0</sub>''</tt> | ||
| + | | : universe name for the dispersed medium | ||
| + | |- | ||
| + | | <tt>''U<sub>bg</sub>''</tt> | ||
| + | | : background universe, i.e. universe filling the space between particles / pebbles | ||
| + | |- | ||
| + | | <tt>''FILE''</tt> | ||
| + | |: input file containing the particle/pebble data | ||
| + | |- | ||
| + | | <tt>''OPT''</tt> | ||
| + | |: additional options (currently only '''pow''' to produce power distribution in a separate output file) | ||
| + | |} | ||
| + | The syntax of the file containing the particle/pebble data is: | ||
| + | |||
| + | ''X<sub>1</sub>'' ''Y<sub>1</sub>'' ''Z<sub>1</sub>'' ''R<sub>1</sub>'' ''U<sub>1</sub>'' | ||
| + | |||
| + | ''X<sub>2</sub>'' ''Y<sub>2</sub>'' ''Z<sub>2</sub>'' ''R<sub>2</sub>'' ''U<sub>2</sub>'' | ||
| + | |||
| + | ... | ||
| + | |||
| + | Where: | ||
| + | {| | ||
| + | |<tt>''X<sub>n</sub>'', ''Y<sub>n</sub>'', ''Z<sub>n</sub>''</tt> | ||
| + | |: are the coordinates | ||
| + | |- | ||
| + | |<tt>''R<sub>n</sub>''</tt> | ||
| + | |: is the radius | ||
| + | |- | ||
| + | |<tt>''U<sub>n</sub>''</tt> | ||
| + | |: is the universe | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Creates a universe (<tt>''U<sub>0</sub>''</tt>), which is filled with spherical sub-universes for which the coordinates are read from a separate file. | ||
| + | *The coordinates can be defined manually, or using the [[Installing_and_running_Serpent#Particle_disperser_routine|automated disperser routine]]. | ||
| + | *Can be used for modelling stochastic particle / pebble-bed geometries in multiple levels. | ||
| + | *If the power distribution option is set, the pebble/particle-wise distribution is written in file <tt>[U<sub>0</sub>].out</tt>. | ||
| + | *See also [[Collection_of_example_input_files#Simple_burnup_examples|HTGR geometry examples]]. | ||
| + | |||
| + | === phb (pulse-height Gaussian energy broadening definition) <span id="phb"></span>=== | ||
| + | |||
| + | '''phb''' ''NAME'' ''TYPE'' [ ... ] | ||
| + | |||
| + | Defines a user-defined (Gaussian) energy broadening function for pulse-height detector ([[#det_dphb|'''dphb''']]). Input values: | ||
| + | |||
| + | {| | ||
| + | |<tt>''NAME''</tt> | ||
| + | |: pulse-height (Gaussian) energy broadening function name | ||
| + | |- | ||
| + | |<tt>''TYPE''</tt> | ||
| + | |: pulse-height function type (1 = energy-resolution interpolation, 2 = energy-FWHM interpolation, 3 = energy-resolution fitting, 4 = energy-FWHM fitting) | ||
| + | |} | ||
| + | |||
| + | The syntax for the different types is as follows: | ||
| + | |||
| + | '''phb''' ''NAME'' '''1''' ''INTT'' ''E<sub>max,1</sub>'' ''R<sub>1</sub>'' ''E<sub>max,2</sub>'' ''R<sub>2</sub>'' ... | ||
| + | |||
| + | where: | ||
| + | {| | ||
| + | |<tt>''INTT''</tt> | ||
| + | |: is the interpolation type (currently only supported type is 2 = lin-lin interpolation data) | ||
| + | |- | ||
| + | |<tt>''E<sub>max,i</sub>, R<sub>i</sub>''</tt> | ||
| + | |: are the maximum energy-resolution tabulated pairs | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | * Full width at half maximum is calculated as: <math> FWHM(E_{max,i}) = R(E_{max,i}) E_{max,i} </math> | ||
| + | * Energies should be given in ascending order. | ||
| + | |||
| + | '''phb''' ''NAME'' '''2''' ''INTT'' ''E<sub>max,1</sub>'' ''FWHM<sub>1</sub>'' ''E<sub>max,2</sub>'' ''FWHM<sub>2</sub>'' ... | ||
| + | |||
| + | where: | ||
| + | {| | ||
| + | |<tt>''INTT''</tt> | ||
| + | |: is the interpolation type (currently only supported type is 2 = lin-lin interpolation data) | ||
| + | |- | ||
| + | |<tt>''E<sub>max,i</sub>, FWHM<sub>i</sub>''</tt> | ||
| + | |: are the maximum energy-full width at half maximum pairs | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | * Energies should be given in ascending order. | ||
| + | |||
| + | '''phb''' ''NAME'' '''3''' ''a'' ''b'' | ||
| + | |||
| + | where: | ||
| + | {| | ||
| + | |<tt>''a, b''</tt> | ||
| + | |: are the parameters to define the energy resolution fit: <math> R = aE^b </math> | ||
| + | |} | ||
| + | |||
| + | '''phb''' ''NAME'' '''4''' ''a'' ''b'' ''c'' | ||
| + | |||
| + | where: | ||
| + | {| | ||
| + | |<tt>''a, b, c''</tt> | ||
| + | |: are the parameters to define the energy full width at half maximum fit: <math> FWHM = a + b\sqrt{(E + cE^2)} </math> | ||
| + | |} | ||
| + | |||
| + | === pin (pin geometry definition)<span id="pin"></span> === | ||
| + | |||
| + | '''pin''' ''U'' | ||
| + | [ ''MAT<sub>1</sub>'' ''R<sub>1</sub>'' ] | ||
| + | [ ''MAT<sub>2</sub>'' ''R<sub>2</sub>'' ] | ||
| + | ... | ||
| + | [ ''MAT<sub>N</sub>'' ] | ||
| + | Defines a pin universe. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''U''</tt> | ||
| + | | : universe name | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub> ... MAT<sub>N</sub>''</tt> | ||
| + | | : material regions | ||
| + | |- | ||
| + | | <tt>''R<sub>1</sub> ... R<sub>N-1</sub>''</tt> | ||
| + | | : outer radii | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The pin card defines an entire universe consisting of nested annular material regions. The boundaries are defined by axially infinite cylindrical surfaces. The outermost region is radially infinite. | ||
| + | *The material entries can be replaced by <tt>'''fill''' ''U<sub>0</sub>''</tt>, in which case the region is filled by another universe. | ||
| + | *Most typically used for defining fuel pins, but can also be applied to guide tubes, control rods, etc. | ||
| + | *The pin card is special case of a [[#nest (nested universe definition)|nested universe type]]. | ||
| + | |||
| + | === plot (geometry plot definition)<span id="plot"></span> === | ||
| + | |||
| + | '''plot''' ''TYPE'' ''XPIX'' ''YPIX'' [ ''POS'' ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ] | ||
| + | |||
| + | '''plot''' ''TYPE'' ''F<sub>min</sub>'' ''F<sub>max</sub>'' ''E'' ''XPIX'' ''YPIX'' [ ''POS'' ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ] | ||
| + | |||
| + | Produces a png-format geometry plot. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : defines the plot type (orientation and plotting of boundaries) | ||
| + | |- | ||
| + | | <tt>''XPIX''</tt> | ||
| + | | : horizontal image size in pixels | ||
| + | |- | ||
| + | | <tt>''YPIX''</tt> | ||
| + | | : vertical image size in pixels | ||
| + | |- | ||
| + | | <tt>''POS''</tt> | ||
| + | | : position of plot plane | ||
| + | |- | ||
| + | | <tt>''MIN<sub>1</sub>''</tt> | ||
| + | | : minimum horizontal coordinate of plotted region | ||
| + | |- | ||
| + | | <tt>''MAX<sub>1</sub>''</tt> | ||
| + | | : maximum horizontal coordinate of plotted region | ||
| + | |- | ||
| + | | <tt>''MIN<sub>2</sub>''</tt> | ||
| + | | : minimum vertical coordinate of plotted region | ||
| + | |- | ||
| + | | <tt>''MAX<sub>2</sub>''</tt> | ||
| + | | : maximum vertical coordinate of plotted region | ||
| + | |- | ||
| + | | <tt>''F<sub>min</sub>''</tt> | ||
| + | | : minimum importance for importance map plots | ||
| + | |- | ||
| + | | <tt>''F<sub>max</sub>''</tt> | ||
| + | | : maximum importance for importance map plots | ||
| + | |- | ||
| + | | <tt>''E''</tt> | ||
| + | | : particle energy for importance map plots | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The <tt>''TYPE''</tt> parameter consists of one or two concatenated values ('AB'): | ||
| + | *#The first value ('A') defines the plot plane (1 = yz, 2 = xz, 3 = xy). | ||
| + | *#The second value ('B') defines which boundaries are plotted (0 = no boundaries, 1 = cell boundaries, 2 = material boundaries, 3 = both). If the second value in is not provided, material boundaries are plotted. | ||
| + | *Importance maps read using the [[#wwin (weight window mesh definition)|wwin card]] can be plotted on top of the geometry by setting the second value ('B') of the type parameter to 4 (linear color scheme) or 5 (logarithmic color scheme) for cell importances, and to 6 (linear color scheme) or 7 (logarithmic color scheme) for source importances. The input parameters then also include the minimum and maximum importance and the particle energy. If importance maps are provided for both neutrons and photons, they can be plotted by entering positive and negative energy values, respectively. | ||
| + | *If both, minimum and maximum importance values are set to "-1", Serpent automatically adjusts them based on the weight-window mesh data, from version 2.2.0 and on. | ||
| + | *Each material plotted with different color. The colors are sampled randomly, unless defined using the '''rgb''' entry in the [[#mat (material definition)|material card]]. | ||
| + | *Void is plotted in black and special colors are used to plot geometry errors (red = overlap, green = undefined region). | ||
| + | *The position parameter defines the location of the plot plane on the axis perpendicular to it (e.g. z-coordinate for xy-type plot). | ||
| + | *The minimum and maximum coordinates define the boundaries of the plotted region (e.g. minimum and maximum x- and y-coordinates for xy-type plot). If these coordinates are not provided, the plot is extended to the maximum dimensions of the geometry. | ||
| + | *The relative dimensions of image size in pixels should match that of the plotted region. Otherwise the image gets distorted. | ||
| + | *Geometry plotter requires compiling the source code with [[Compiling Serpent|GD Graphics libraries]]. | ||
| + | *[[Installing and running Serpent#Running Serpent|Command line parameter]] <tt>''-plot''</tt> stops the execution after the geometry plots are produced. Option <tt>''-qp''</tt> invokes a quick plot mode, which does not check for overlaps. Option <tt>''-noplot''</tt> skips the geometry plots altogether. | ||
| + | *See also [[Visualizing the results#Geometry plotter|detailed description]] on geometry plotter. | ||
| + | *Geometry plot produced by the nth plot-card is written in file <tt>[input]_geom[n].png</tt>. | ||
| + | *''Note to developers: particle type should be included as an input parameter in importance map plots.'' | ||
| + | |||
| + | === rep (reprocessor definition)<span id="rep"></span> === | ||
| + | |||
| + | '''rep''' ''NAME'' | ||
| + | [ '''rc''' ''SRC'' ''TGT'' ''MFLOW'' ''MODE'' ] | ||
| + | [ '''rm''' ''MAT<sub>1</sub>'' ''MAT<sub>2</sub>'' ] | ||
| + | [ '''ru''' ''UNI<sub>1</sub>'' ''UNI<sub>2</sub>'' ] | ||
| + | |||
| + | Defines the reprocessing controllers. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : name of the reprocessor. | ||
| + | |- | ||
| + | | <tt>''SRC''</tt> | ||
| + | | : name of the source material, from which the flow is moved | ||
| + | |- | ||
| + | | <tt>''TGT''</tt> | ||
| + | | : name of the target material, to which the flow is moved | ||
| + | |- | ||
| + | | <tt>''MFLOW''</tt> | ||
| + | | : name of the material flow | ||
| + | |- | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : continuous reprocessing mode | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub>''</tt> | ||
| + | | : name of the replaced material | ||
| + | |- | ||
| + | | <tt>''MAT<sub>2</sub>''</tt> | ||
| + | | : name of the replacing material | ||
| + | |- | ||
| + | | <tt>''UNI<sub>1</sub>''</tt> | ||
| + | | : name of the replaced universe | ||
| + | |- | ||
| + | | <tt>''UNI<sub>2</sub>''</tt> | ||
| + | | : name of the replacing universe | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The reprocessor name identifies the reprocessing regime in the depletion calculation [[#dep|dep card]]. The syntax corresponds to '''dep pro''' ''NAME''. | ||
| + | *The nuclides identifier of those included in both source <tt>''SRC''</tt> and target <tt>''TGT''</tt> materials in reprocessors should follow the same format, either ZA.ID or ISO.ID (for nuclides with cross sections), or ZAI (for nuclides without associated cross sections, and adding the '''fix''' entry to the [[#mat (material definition)|mat card]]). | ||
| + | *The (continuous) reprocessing implementation works with materials, not universes. Therefore, define the universes associated with those burnable materials as surface-cell type universes. | ||
| + | *Multiple reprocessing controllers/regimes can be defined within the same reprocessor definition. | ||
| + | *The '''rc''' continuous reprocessing option can be used to define the material flow between the source and the target materials. | ||
| + | **The material flow is defined using the [[#mflow|mflow card]]. | ||
| + | **The continuous reprocessing <tt>''MODE''</tt> defines how to incorporate the material flow into the Bateman equations: | ||
| + | |||
| + | ::{|class="wikitable" style="text-align: center;" | ||
| + | ! <tt>''MODE''</tt> | ||
| + | ! Material source | ||
| + | ! Material target | ||
| + | ! Material flow | ||
| + | |- | ||
| + | | 0 | ||
| + | | <math>N_{src}(t)=N_{src}(0)</math> | ||
| + | | <math>N_{tgt}(t)=N_{tgt}(0)+t\lambda N_{src}(0)</math> | ||
| + | | <math>\dot{N}(t)=\lambda N_{src}(0)</math> | ||
| + | |- | ||
| + | | 1 | ||
| + | | <math>N_{src}(t)=N_{src}(0)e^{-\lambda t}</math> | ||
| + | | <math>N_{tgt}(t) = N_{tgt}(0) + (1-e^{-\lambda t})N_{src}(0)</math> | ||
| + | | <math>\dot{N}(t)=\lambda N_{src}(t)</math> | ||
| + | |- | ||
| + | | 2 | ||
| + | | <math>N_{src}(n+1)=(1-\Delta t_{n}\lambda)N_{src}(n)</math> | ||
| + | | <math>N_{tgt}(n+1)=N_{tgt}(n)+\Delta t_{n}\lambda N_{src}(n)</math> | ||
| + | | <math>\dot{N}(n)=\lambda N_{src}(n)</math> | ||
| + | |} | ||
| + | |||
| + | :*<tt>''MODE''</tt> 0 : no changes at the source material and adds ''λN<sub>0</sub>'' from the source material to the target material when solving the Bateman equations (''N<sub>0</sub>'' are initial compositions). | ||
| + | :*<tt>''MODE''</tt> 1 : subtracts ''λN'' from the source material and adds it to the target material when solving the Bateman equations. | ||
| + | :*<tt>''MODE''</tt> 2 : subtracts ''λN<sub>n</sub>'' from the source material and adds it to the target material when solving the Bateman equations (compositions updated with each burnup step, ''n''). | ||
| + | *The '''rm''' material reprocessing option replaces one material with another, ''MAT<sub>1</sub>'' by ''MAT<sub>2</sub>''. | ||
| + | *The '''ru''' universe reprocessing option replaces one universe with another, ''UNI<sub>1</sub>'' by ''UNI<sub>2</sub>''. | ||
| + | |||
| + | === sample (Temperature / density data sample definition)<span id="sample"></span> === | ||
| + | |||
| + | '''sample''' ''N<sub>X</sub>'' ''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ''N<sub>Y</sub>'' ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ''N<sub>Z</sub>'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' | ||
| + | |||
| + | Samples values from the initial material temperatures and densities to a file using a Cartesian grid. | ||
| + | |||
| + | Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : Number of values to sample in the x-direction. | ||
| + | |- | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : Minimum coordinate to sample from in the x-direction. | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : Maximum coordinate to sample from in the x-direction. | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : Number of values to sample in the y-direction. | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : Minimum coordinate to sample from in the y-direction. | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : Maximum coordinate to sample from in the y-direction. | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : Number of values to sample in the z-direction. | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : Minimum coordinate to sample from in the z-direction. | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : Maximum coordinate to sample from in the z-direction. | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The data from each sample is written in a separate <tt>[input]_sample''N''.m</tt> file. | ||
| + | *Positive values for the density data correspond to atomic densities, while negative values correspond to mass densities. | ||
| + | *Materials with no temperature specified either in their [[Input syntax manual#mat|mat]]-card or through an interface definition will show a temperature of 0. | ||
| + | |||
| + | === sens (sensitivity calculation definition)<span id="sens"></span> === | ||
| + | '''sens''' '''pert''' | ||
| + | |||
| + | '''sens''' '''resp''' | ||
| + | |||
| + | '''sens''' '''opt''' | ||
| + | |||
| + | Definitions for the perturbations, responses and options for [[sensitivity calculations]]. | ||
| + | |||
| + | === solid (irregular 3D geometry definition)<span id="solid"></span> === | ||
| + | '''solid 1''' ''UNI'' ''BGUNI'' | ||
| + | ''MESH_SPLIT'' ''MESH_DIM'' ''SZ<sub>1</sub>'' ''SZ<sub>2</sub>'' ... ''SZ<sub>MESH_DIM</sub>'' | ||
| + | ''POINTS_FILE'' | ||
| + | ''FACES_FILE'' | ||
| + | ''OWNER_FILE'' | ||
| + | ''NEIGHBOUR_FILE'' | ||
| + | ''MATERIALS_FILE'' | ||
| + | |||
| + | Creates an unstructured mesh-based geometry universe. Input values are: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name for the irregular geometry | ||
| + | |- | ||
| + | | <tt>''BGUNI''</tt> | ||
| + | | : name of the background universe filling all undefined space | ||
| + | |- | ||
| + | | <tt>''MESH_SPLIT''</tt> | ||
| + | | : Splitting criterion for the adaptive search mesh (maximum number of geometry cells in search mesh cell) | ||
| + | |- | ||
| + | | <tt>''MESH_DIM''</tt> | ||
| + | | : number of levels in the adaptive search mesh | ||
| + | |- | ||
| + | | <tt>''SZ<sub>i</sub>''</tt> | ||
| + | | : Size of the search mesh at level <tt>''i''</tt> | ||
| + | |- | ||
| + | | <tt>''POINTS_FILE''</tt> | ||
| + | | : Path to the unstructured mesh points file | ||
| + | |- | ||
| + | | <tt>''FACES_FILE''</tt> | ||
| + | | : Path to the unstructured mesh faces file | ||
| + | |- | ||
| + | | <tt>''OWNER_FILE''</tt> | ||
| + | | : Path to the unstructured mesh owner file | ||
| + | |- | ||
| + | | <tt>''NEIGHBOUR_FILE''</tt> | ||
| + | | : Path to the unstructured mesh neighbour file | ||
| + | |- | ||
| + | | <tt>''MATERIALS_FILE''</tt> | ||
| + | | : Path to the unstructured mesh materials file | ||
| + | |} | ||
| + | |||
| + | '''solid 2''' ''UNI'' ''BGUNI'' | ||
| + | ''MESH_SPLIT'' ''MESH_DIM'' ''SZ<sub>1</sub>'' ''SZ<sub>2</sub>'' ... ''SZ<sub>MESH_DIM</sub>'' | ||
| + | ''MODE'' ''R0'' | ||
| + | '''body''' ''BODY<sub>1</sub>'' ''CELL<sub>1</sub>'' ''MAT<sub>1</sub>'' | ||
| + | '''file''' ''BODY<sub>1</sub>'' ''FILE<sub>1</sub>'' ''SCALE<sub>1</sub>'' ''X<sub>1</sub>'' ''Y<sub>1</sub>'' ''Z<sub>1</sub>'' | ||
| + | '''file''' ''BODY<sub>1</sub>'' ''FILE<sub>2</sub>'' ''SCALE<sub>2</sub>'' ''X<sub>2</sub>'' ''Y<sub>2</sub>'' ''Z<sub>2</sub>'' | ||
| + | ... | ||
| + | '''body''' ''BODY<sub>2</sub>'' ''CELL<sub>2</sub>'' ''MAT<sub>2</sub>'' | ||
| + | '''file''' ''BODY<sub>2</sub>'' ''FILE<sub>3</sub>'' ''SCALE<sub>3</sub>'' ''X<sub>3</sub>'' ''Y<sub>3</sub>'' ''Z<sub>3</sub>'' | ||
| + | '''file''' ''BODY<sub>2</sub>'' ''FILE<sub>4</sub>'' ''SCALE<sub>4</sub>'' ''X<sub>4</sub>'' ''Y<sub>4</sub>'' ''Z<sub>4</sub>'' | ||
| + | ... | ||
| + | |||
| + | Creates an STL-based geometry universe. Input values are: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name for the irregular geometry | ||
| + | |- | ||
| + | | <tt>''BGUNI''</tt> | ||
| + | | : name of the background universe filling all undefined space | ||
| + | |- | ||
| + | | <tt>''MESH_SPLIT''</tt> | ||
| + | | : Splitting criterion for the adaptive search mesh (maximum number of geometry cells in search mesh cell) | ||
| + | |- | ||
| + | | <tt>''MESH_DIM''</tt> | ||
| + | | : number of levels in the adaptive search mesh | ||
| + | |- | ||
| + | | <tt>''SZ<sub>i</sub>''</tt> | ||
| + | | : Size of the search mesh at level <tt>''i''</tt> | ||
| + | |- | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : Mode for handling the triangulated geometry (1 = "fast", 2 = "safe"). | ||
| + | |- | ||
| + | | <tt>''R0''</tt> | ||
| + | | : Radius inside which two points of the STL-geometry are joined into one. | ||
| + | |- | ||
| + | | <tt>''BODY<sub>i</sub>''</tt> | ||
| + | | : Name of solid body <tt>''i''</tt> | ||
| + | |- | ||
| + | | <tt>''CELL<sub>i</sub>''</tt> | ||
| + | | : Name of geometry cell <tt>''i''</tt> linked with body <tt>''i''</tt> | ||
| + | |- | ||
| + | | <tt>''MAT<sub>i</sub>''</tt> | ||
| + | | : Material filling cell <tt>''i''</tt> | ||
| + | |- | ||
| + | | <tt>''FILE<sub>i</sub>''</tt> | ||
| + | | : Path to a file containing an STL solid model, multiple files can be linked to one body | ||
| + | |- | ||
| + | | <tt>''SCALE<sub>i</sub>''</tt> | ||
| + | | : Scaling factor for the STL model in <tt>''FILE<sub>i</sub>''</tt> | ||
| + | |- | ||
| + | | <tt>''X<sub>i</sub>''</tt> | ||
| + | | : Shift in x-direction to the STL model in <tt>''FILE<sub>i</sub>''</tt> | ||
| + | |- | ||
| + | | <tt>''Y<sub>i</sub>''</tt> | ||
| + | | : Shift in y-direction to the STL model in <tt>''FILE<sub>i</sub>''</tt> | ||
| + | |- | ||
| + | | <tt>''Z<sub>i</sub>''</tt> | ||
| + | | : Shift in z-direction to the STL model in <tt>''FILE<sub>i</sub>''</tt> | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The material entries can be replaced by <tt>'''fill''' ''UNI<sub>i</sub>''</tt>, in which case the cell <tt>''i''</tt> is filled by the given universe. | ||
| + | |||
| + | '''solid 3''' | ||
| + | ''INTERFACE_FILE'' | ||
| + | |||
| + | Creates an unstructured mesh-based geometry universe with unstructured mesh-based temperature and/or density distributions. Input values are: | ||
| + | |||
| + | {| | ||
| + | | <tt>''INTERFACE_FILE''</tt> | ||
| + | | : Path to the [[Multi-physics_interface#Unstructured_mesh_based_interface_and_geometry_definition_.28type_9.29|interface file]] containing the rest of the parameters | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *For simple example of a CAD-based geometry, see the [[Stanford critical bunny]]. | ||
| + | *For more information on the unstructured mesh based geometry see [[Unstructured mesh based input]]. | ||
| + | *For a simple example of an unstructured mesh based geometry see the [[Simple umsh 8 cubes input]]. | ||
| + | |||
| + | === src (source definition)<span id="src"></span> === | ||
| + | |||
| + | '''src''' ''NAME'' [ ''PART'' ] | ||
| + | [ [[#src_sw|'''sw''']] ''WGT'' ] | ||
| + | [ [[#src_sc|'''sc''']] ''CELL'' ] | ||
| + | [ [[#src_su|'''su''']] ''UNI'' ] | ||
| + | [ [[#src_sm|'''sm''']] ''MAT'' ] | ||
| + | [ [[#src_sp|'''sp''']] ''X'' ''Y'' ''Z'' ] | ||
| + | [ [[#src_sx|'''sx''']] ''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ] | ||
| + | [ [[#src_sy|'''sy''']] ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ] | ||
| + | [ [[#src_sz|'''sz''']] ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ] | ||
| + | [ [[#src_srad|'''srad''']] ''R<sub>MIN</sub>'' ''R<sub>MAX</sub>'' ] | ||
| + | [ [[#src_ss|'''ss''']] ''SURF'' ] | ||
| + | [ [[#src_sd|'''sd''']] ''U'' ''V'' ''W'' ] | ||
| + | [ [[#src_se|'''se''']] ''E'' ] | ||
| + | [ [[#src_sb|'''sb''']] ''N'' ''INTT'' ''E<sub>1</sub>'' ''WGT<sub>1</sub>'' ''E<sub>2</sub>'' ''WGT<sub>2</sub>'' ... ] | ||
| + | [ [[#src_sr|'''sr''']] ''NUC'' ''MT'' ] | ||
| + | [ [[#src_st|'''st''']] ''T<sub>MIN</sub>'' ''T<sub>MIN</sub>'' ] | ||
| + | [ [[#src_sf|'''sf''']] ''FILE'' ''TYPE'' ] | ||
| + | [ [[#src_si|'''si''']] ''N'' ''P<sub>1</sub>'' ''P<sub>2</sub>'' ... ] | ||
| + | [ [[#src_sg|'''sg''']] ''MAT'' ''MODE'' ] | ||
| + | Source definition. The first parameter: | ||
| + | |||
| + | {| | ||
| + | | <tt>''PART''</tt> | ||
| + | | : particle type (n = neutron, p = photon) | ||
| + | |} | ||
| + | |||
| + | is optional in single particle simulations. The remaining parameters are defined by separate key words followed by the input values. | ||
| + | |||
| + | |||
| + | Source weight (<tt>'''sw'''</tt>):<span id="src_sw"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''WGT''</tt> | ||
| + | | : relative source weight | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *When multiple sources are defined, each definition is sampled with equal probability. This probability can be changed by assigning different weights for each source. | ||
| + | *The weights are automatically normalized before the calculation is started. | ||
| + | |||
| + | |||
| + | Source cell (<tt>'''sc'''</tt>):<span id="src_sc"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''CELL''</tt> | ||
| + | | : cell inside which the source points are sampled | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Setting a source cell is one of the options that can be applied to define the spatial distribution of source particles. | ||
| + | *The selection is based on rejection sampling, and if the source cell occupies a small volume of the geometry, the sampling efficiency can be increased by defining a bounding box/(vertical) cylinder around the cell (using the <tt>'''sx'''</tt>, <tt>'''sy'''</tt> and <tt>'''sz'''</tt> or <tt>'''sp'''</tt>, <tt>'''srad'''</tt> and <tt>'''sz'''</tt> options, respectively). | ||
| + | *If no spatial distribution is defined, particles are sampled uniformly over the geometry. | ||
| + | |||
| + | |||
| + | Source universe (<tt>'''su'''</tt>):<span id="src_su"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe inside which the source points are sampled | ||
| + | |} | ||
| + | |||
| + | |||
| + | Source material (<tt>'''sm'''</tt>):<span id="src_sm"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material inside which the source points are sampled | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Setting a source material is one of the options that can be applied to define the spatial distribution of source particles. | ||
| + | *The selection is based on rejection sampling, and if the source material occupies a small volume of the geometry, the sampling efficiency can be increased by defining a bounding box/(vertical) cylinder around the cell (using the <tt>'''sx'''</tt>, <tt>'''sy'''</tt> and <tt>'''sz'''</tt> or <tt>'''sp'''</tt>, <tt>'''srad'''</tt> and <tt>'''sz'''</tt> options, respectively). | ||
| + | *If no spatial distribution is defined, particles are sampled uniformly over the geometry. | ||
| + | |||
| + | |||
| + | Source point (<tt>'''sp'''</tt>):<span id="src_sp"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''X''</tt>, <tt>''Y''</tt>, <tt>''Z''</tt>, | ||
| + | | : coordinates of the source point | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Setting a point source is one of the options that can be applied to define the spatial distribution of source particles. | ||
| + | *If no spatial distribution is defined, particles are sampled uniformly over the geometry. | ||
| + | |||
| + | |||
| + | Source boundaries (<tt>'''sx'''</tt>, <tt>'''sy'''</tt>, <tt>'''sz'''</tt> and <tt>'''srad'''</tt>):<span id="src_sy"></span><span id="src_sx"></span><span id="src_sz"></span><span id="src_srad"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''X<sub>MIN</sub>''</tt>, <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : Boundaries on X-axis | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt>, <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : Boundaries on Y-axis | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt>, <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : Boundaries on Z-axis | ||
| + | |- | ||
| + | | <tt>''R<sub>MIN</sub>''</tt>, <tt>''R<sub>MAX</sub>''</tt> | ||
| + | | : Radial boundaries | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Source boundaries are used to define a bounding box/(vertical) cylinder inside which the source particles are sampled. | ||
| + | *The radial boundaries are centered around the point defined by <tt>'''sp'''</tt> and can be used in combination with <tt>'''sz'''</tt>. | ||
| + | *Can be used in combination with cell and material sources to increase the sampling efficiency. | ||
| + | *If no bounding box is defined, particles are sampled uniformly over the geometry. | ||
| + | |||
| + | |||
| + | Surface source (<tt>'''ss'''</tt>):<span id="src_ss"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''SURF''</tt> | ||
| + | | : surface on which the source particles are sampled | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The surface source is currently limited to infinite vertical cylinder (<tt>'''cyl'''</tt>) and sphere (<tt>'''sph'''</tt>) surface types. | ||
| + | *Particles are started in the direction of the inward surface normal. | ||
| + | |||
| + | |||
| + | Source direction (<tt>'''sd'''</tt>):<span id="src_sd"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''U''</tt>, <tt>''V''</tt>, <tt>''W''</tt>, | ||
| + | | : direction vector of source particles | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The source direction option can be set to define a unidirectional source. | ||
| + | *If no directional dependence is defined, the direction of source particles is sampled isotropically. | ||
| + | |||
| + | |||
| + | Source energy (<tt>'''se'''</tt>):<span id="src_se"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''E''</tt> | ||
| + | | : energy of source particles | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The source energy option can be used to define a monoenergetic source. | ||
| + | *The default energy of neutrons and photons is 1 MeV. | ||
| + | *This option can also be used together with the source reaction option (<tt>'''sr'''</tt>). | ||
| + | |||
| + | |||
| + | Source energy bins (<tt>'''sb'''</tt>):<span id="src_sb"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''N''</tt> | ||
| + | | : number of bins | ||
| + | |- | ||
| + | | <tt>''INTT''</tt> | ||
| + | | : interpolation (0 = line spectrum, 1 = histogram, 2 = lin-lin, 4 = log-lin) | ||
| + | |- | ||
| + | | <tt>''E<sub>n</sub>''</tt> | ||
| + | | : upper boundary of the energy bin | ||
| + | |- | ||
| + | | <tt>''WGT<sub>n</sub>''</tt> | ||
| + | | : weight of the energy bin | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option allows defining an arbitrary source spectrum in the form of tabular data. | ||
| + | *The bins are entered in the order of ascending energy, and weight of the first bin must be set to zero. | ||
| + | *Interpolation is given in a separate parameter from version 2.1.31 on. | ||
| + | *Here, a simple test input that demonstrates the [[Source energy spectrum definition test case|source spectrum definition]]. | ||
| + | |||
| + | Source reaction (<tt>'''sr'''</tt>):<span id="src_sr"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''NUC''</tt> | ||
| + | | : nuclide name | ||
| + | |- | ||
| + | | <tt>''MT''</tt> | ||
| + | | : reaction number | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The source reaction determines a distribution function for source energy (for example, <sup>235</sup>U fission spectrum can be defined as: <tt>'''sr''' ''92235.09c'' ''18''</tt>). | ||
| + | *The reaction numbers are [[ENDF reaction MT's and macroscopic reaction numbers|ENDF reaction MT's]], and the data is obtained from standard cross section libraries. | ||
| + | *Applies to neutrons only. | ||
| + | *When the source energy parameter (<tt>'''se'''</tt>) is defined, the value is used as the energy of the incoming neutrons. | ||
| + | |||
| + | |||
| + | Source time (<tt>'''st'''</tt>):<span id="src_st"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''T<sub>MIN</sub>''</tt>, <tt>''T<sub>MAX</sub>''</tt> | ||
| + | | : time boundaries | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This parameter defines a time interval for the sampled source particles. The starting time is sampled uniformly between the given minimum and maximum. | ||
| + | *All source particles are started at time zero by default. | ||
| + | |||
| + | |||
| + | Source file (<tt>'''sf'''</tt>):<span id="src_sf"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : file path to source file | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : file type (-1 = binary, 1 = ASCII) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Source files allow defining arbitrary distributions by reading the particle coordinates, direction, energy, weight and time from a file. | ||
| + | *Source files can be produced using the <tt>'''df'''</tt> entry of [[#det_df|detector cards]], or the [[#set csw|set csw]] or [[#set gsw|set gsw]] options. | ||
| + | |||
| + | |||
| + | User-defined source routine (<tt>'''si'''</tt>):<span id="src_si"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''N''</tt> | ||
| + | | : number of parameters | ||
| + | |- | ||
| + | | <tt>''P<sub>n</sub>''</tt> | ||
| + | | : parameters passed as arguments into the subroutine | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option allows defining an arbitrary source distributions with a user-defined subroutine. | ||
| + | *The source parameters are passed as arguments into the subroutine, together, with sampled position, direction energy, weight and time. | ||
| + | *For complete description see source file "<tt>usersrc.c</tt>". | ||
| + | *The subroutine may be overwritten with the blank template file when installing updates. | ||
| + | |||
| + | |||
| + | Radioactive decay source (<tt>'''sg'''</tt>):<span id="src_sg"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material name or -1 | ||
| + | |- | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : sampling mode (1 = analog, 2 = implicit) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Radioactive decay source combines material compositions to decay data read from ENDF format libraries and forms the normalized source distribution automatically. | ||
| + | *Material compositions can be defined manually, or read from binary restart files produced by a burnup or activation calculation (see the [[#set rfw|set rfw]] and [[#set rfr|set rfr]] options). | ||
| + | *If the material name is replaced by -1, source points are started from all radioactive materials. | ||
| + | *The analog sampling mode preserves the average number of particles produced in radioactive decay, but may lead to poor sampling efficiency in geometries with both low and high-active materials. | ||
| + | *The implicit sampling mode preserves the total statistical weight of emitted particles and produces a uniform source distribution over activated materials. | ||
| + | *In version 2.1.28 the source is limited to photon line spectra. | ||
| + | *The calculation produces an additional output file <tt>[input]_gsrc.m</tt> or <tt>[input]_nsrc.m</tt> that contains the gamma/neutron source spectra, respectively. | ||
| + | *See [[Radioactive decay source, practical example|practical example]] for more information. | ||
| + | |||
| + | === srtrans (source transformation)<span id="srtrans"></span> === | ||
| + | |||
| + | See [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === strans (surface transformation)<span id="strans"></span> === | ||
| + | |||
| + | See [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === surf (surface definition)<span id="surf"></span> === | ||
| + | |||
| + | '''surf''' ''NAME TYPE'' [ ''PARAM<sub>1</sub> PARAM<sub>2</sub>'' ... ] | ||
| + | |||
| + | Defines a surface. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : is the surface name | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : is the surface type | ||
| + | |- | ||
| + | | <tt>''PARAM<sub>n</sub>''</tt> | ||
| + | | : are the surface parameters | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The name is used to identify the surface, for example, in the [[#cell (cell definition)|cell card]]. | ||
| + | *See [[Surface types|separate description on surface types]]. | ||
| + | *Surfaces can be moved and rotated using [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === therm and thermstoch (thermal scattering)<span id="therm"></span><span id="thermstoch"></span> === | ||
| + | |||
| + | '''therm''' ''NAME'' ''LIB'' | ||
| + | |||
| + | '''therm''' ''NAME'' ''TEMP'' ''LIB<sub>1</sub>'' ''LIB<sub>2</sub>'' | ||
| + | |||
| + | '''therm''' ''NAME'' 0 ''LIB<sub>1</sub>'' ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... | ||
| + | |||
| + | '''thermstoch''' ''NAME'' ''TEMP'' ''LIB<sub>1</sub>'' ''LIB<sub>2</sub>'' | ||
| + | |||
| + | Defines thermal scattering data that can be linked to nuclides using input entry '''moder''' in the [[#mat (material definition)|material cards]]. When using thermal scattering data together with TMS on-the-fly temperature treatment, the third input value of the therm card is 0. In this case, Serpent interpolates the thermal scattering data automatically to the local temperature, as defined either using the '''tms''' input entry in the material definition ([[#mat (material definition)|mat card]]) or via the multi-physics interface ([[#ifc (interface file)|ifc card]]). | ||
| + | |||
| + | Input values: | ||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : name of the thermal scattering data | ||
| + | |- | ||
| + | | <tt>''LIB<sub>i</sub>''</tt> | ||
| + | | : thermal scattering data identifiers as defined in the directory file (acelib) | ||
| + | |- | ||
| + | | <tt>''TEMP''</tt> | ||
| + | | : temperature to which the thermal scattering data is interpolated | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *When using on-the-fly interpolation of thermal scattering data, <tt>''LIB<sub>i</sub>''</tt> must cover the whole temperature range in which the materials appear in the geometry. I.e. extrapolation of the data is not supported. | ||
| + | *Thermal scattering data is interpolated using the methodology of makxsf code | ||
| + | *The interpolation can be performed using the stochastic mixing approach with '''thermstoch'''. This interpolation mode is not available for on-the-fly interpolation. | ||
| + | *The continuous S(α, β) formalism is available from version 2.1.32 on. | ||
| + | *Version 2.2.0 includes the on-the-fly temperature treatment for the continuous S(α,β) formalism. | ||
| + | |||
| + | === tme (time binning definition)<span id="tme"></span> === | ||
| + | |||
| + | '''tme''' ''NAME'' '''1''' ''LIM<sub>1</sub> LIM<sub>2</sub>'' ... | ||
| + | |||
| + | '''tme''' ''NAME'' '''2''' ''NB T<sub>min</sub> T<sub>max</sub>'' | ||
| + | |||
| + | '''tme''' ''NAME'' '''3''' ''NB T<sub>min</sub> T<sub>max</sub>'' | ||
| + | |||
| + | Defines a time binning structure. The second entry sets the binning type (1 = arbitrary, 2 = uniform, 3 = log-uniform). Remaining values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : name of the time binning | ||
| + | |- | ||
| + | | <tt>''NB''</tt> | ||
| + | | : number of bins | ||
| + | |- | ||
| + | | <tt>''LIM<sub>n</sub>''</tt> | ||
| + | | : time bin boundaries in arbitrary binning | ||
| + | |- | ||
| + | | <tt>''T<sub>min</sub>''</tt> | ||
| + | | : minimum time boundary in uniform or log-uniform binning | ||
| + | |- | ||
| + | | <tt>''T<sub>max</sub>''</tt> | ||
| + | | : maximum time boundary in uniform or log-uniform binning | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The entered values are in seconds | ||
| + | *The first limit in the arbitrary type (type = 1), is the lower bound of the first bin. The second limit is the upper bound of the first bin and so on. | ||
| + | *Time binning is used with [[#det (detector definition)|detectors]] and [[Dynamic external source simulation mode|dynamic simulation mode]]. | ||
| + | |||
| + | === trans (transformations)<span id="trans"></span> === | ||
| + | |||
| + | '''trans''' ''TYPE'' ''UNIT'' [ ''IDX'' ] ''LVL'' | ||
| + | |||
| + | '''trans''' ''TYPE'' ''UNIT'' [ ''IDX'' ] ''X'' ''Y'' ''Z'' | ||
| + | |||
| + | '''trans''' ''TYPE'' ''UNIT'' [ ''IDX'' ] ''X'' ''Y'' ''Z'' ''θ<sub>x</sub>'' ''θ<sub>y</sub>'' ''θ<sub>z</sub>'' ''ORD'' | ||
| + | |||
| + | '''trans''' ''TYPE'' ''UNIT'' [ ''IDX'' ] ''X'' ''Y'' ''Z'' ''α<sub>1</sub>'' ''α<sub>2</sub>'' ''α<sub>3</sub>'' ''α<sub>4</sub>'' ''α<sub>5</sub>'' ''α<sub>6</sub>'' ''α<sub>7</sub>'' ''α<sub>8</sub>'' ''α<sub>9</sub>'' ''ORD'' | ||
| + | |||
| + | '''trans''' ''TYPE'' ''UNIT'' [ ''IDX'' ] '''rot''' ''X<sub>0</sub>'' ''Y<sub>0</sub>'' ''Z<sub>0</sub>'' ''I'' ''J'' ''K'' ''β'' | ||
| + | |||
| + | Defines surface, universe, fill, lattice, detector mesh or source transformation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : type of transformation (S = surface, F = fill, U = universe, L = lattice, D = detector mesh, SR = source) | ||
| + | |- | ||
| + | | <tt>''UNIT''</tt> | ||
| + | | : surface, cell, universe, lattice, detector mesh or source name to which the transformation is applied | ||
| + | |- | ||
| + | | <tt>''IDX''</tt> | ||
| + | | : index number in lattice transformation (type L) | ||
| + | |- | ||
| + | | <tt>''LVL''</tt> | ||
| + | | : level number in universe level transformation | ||
| + | |- | ||
| + | | <tt>''X'',''Y'',''Z''</tt> | ||
| + | | : translation vector | ||
| + | |- | ||
| + | | <tt>''θ<sub>x</sub>'' ''θ<sub>y</sub>'' ''θ<sub>z</sub>''</tt> | ||
| + | | : rotation angles with respect to x-, y- and z-axes (in degrees) | ||
| + | |- | ||
| + | | <tt>''α<sub>1</sub>'' ... ''α<sub>9</sub>''</tt> | ||
| + | | : coefficients of the rotation matrix | ||
| + | |- | ||
| + | | <tt>''ORD''</tt> | ||
| + | | : order in which translations and rotations are applied (1 = rotations first, 2 = translations first) | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>'',''Y<sub>0</sub>'',''Z<sub>0</sub>''</tt> | ||
| + | | : Origin of vector defining rotation axis. | ||
| + | |- | ||
| + | | <tt>''I'',''J'',''K''</tt> | ||
| + | | : Components of vector defining rotation axis. | ||
| + | |- | ||
| + | | <tt>''β''</tt> | ||
| + | | : Angle around rotation axis defined by a vector (in degrees). '''NB: In Serpent 2.1.29 positive values correspond to rotation to the negative mathematical direction and vice versa.''' | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Fill transformation is applied in the universe filling the given cell. | ||
| + | *Level transformation is a special type of universe transformation, in which the coordinates in the given universe are obtained relative to geometry level <tt>''LVL''</tt>. | ||
| + | *Lattice transformation requires to provide the index for the transformation <tt>''IDX''</tt>. | ||
| + | *Source transformation is inverted. transformation is inverted compared to how surface, universe, etc. are handled. | ||
| + | *By default translations are applied before rotations, and the order can be switched using the <tt>''ORD''</tt> parameter. | ||
| + | *Rotations can be defined either by providing the three angles with respect to the three coordinate axes, or by defining the rotation matrix. In the second case Serpent applies vector multiplication <math>\vec{r'} = \bold{A} \vec{r}</math> where <math>\vec{r}</math> and <math>\vec{r'}</math> are the position vectors before and after the operation and coefficients <tt>''α<sub>1</sub>'' ... ''α<sub>9</sub>''</tt> define the 3 by 3 matrix <math>\bold{A}</math>. | ||
| + | *To preserve backwards compatibility, input parameters "strans", "utrans", "ftrans", "ltrans", "dtrans" and "srtrans" without the following type identifier are also accepted for defining surface, universe, fill, lattice, detector mesh and source transformations, respectively. To preserve compatibility with Serpent 1, parameter "trans" without type identifier defines a universe transformation. | ||
| + | |||
| + | === transb (burnup transformation)<span id="transb"></span> === | ||
| + | |||
| + | '''transb''' ''STEP'' [ <''trans''> ] | ||
| + | |||
| + | Defines burnup-dependent surface, universe or fill transformation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''STEP''</tt> | ||
| + | | : step in units of burnup (positive values) or days (negative values) | ||
| + | |- | ||
| + | | <tt><''trans''></tt> | ||
| + | | : list of parameters associated with the transformation | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The parameters associated with the transformation follow the standard transformation cards syntax without "trans" identifier. | ||
| + | |||
| + | === transv and transa (velocity and acceleration transformations)<span id="transa"></span><span id="transv"></span> === | ||
| + | |||
| + | '''transv''' ''TYPE'' ''UNIT'' [ ''IDX'' ] [ '''tlim''' ''T<sub>0</sub>'' ''T<sub>1</sub>'' ''T<sub>TYPE</sub>'' ] ''V<sub>X</sub>'' ''V<sub>Y</sub>'' ''V<sub>Z</sub>'' | ||
| + | |||
| + | '''transa''' ''TYPE'' ''UNIT'' [ ''IDX'' ] [ '''tlim''' ''T<sub>0</sub>'' ''T<sub>1</sub>'' ''T<sub>TYPE</sub>'' ] ''A<sub>X</sub>'' ''A<sub>Y</sub>'' ''A<sub>Z</sub>'' | ||
| + | |||
| + | |||
| + | Defines surface, universe, fill, lattice, detector mesh or source transformation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : type of transformation (S = surface, F = fill, U = universe, L = lattice, D = detector mesh, SR = source) | ||
| + | |- | ||
| + | | <tt>''UNIT''</tt> | ||
| + | | : surface, cell, universe, lattice, detector mesh or source name to which the transformation is applied | ||
| + | |- | ||
| + | | <tt>''IDX''</tt> | ||
| + | | : index number in lattice transformation (type L) | ||
| + | |- | ||
| + | | <tt>''T<sub>0</sub>''</tt> | ||
| + | | : beginning time of the transformation | ||
| + | |- | ||
| + | | <tt>''T<sub>1</sub>''</tt> | ||
| + | | : end time of the transformation | ||
| + | |- | ||
| + | | <tt>''T<sub>TYPE</sub>''</tt> | ||
| + | | : transformation type after end time (1 = movement stops, 2 = transformation removed, 3 = initial acceleration and velocity removed, but velocity accumulated due to acceleration remains) | ||
| + | |- | ||
| + | | <tt>''V<sub>X</sub>'',''V<sub>Y</sub>'',''V<sub>Z</sub>''</tt> | ||
| + | | : Initial velocity vector | ||
| + | |- | ||
| + | | <tt>''A<sub>X</sub>'',''A<sub>Y</sub>'',''A<sub>Z</sub>''</tt> | ||
| + | | : Initial acceleration vector | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Fill transformation is applied in the universe filling the given cell. | ||
| + | *The transformation is updated at the simulation time-interval boundaries. | ||
| + | *See [[UGM 2016 Moving geometry]]. | ||
| + | *See [[Rotating Translating STL Bunny]]. | ||
| + | |||
| + | ===umsh (unstructured mesh-based geometry definition)<span id="transa"></span><span id="umsh"></span> === | ||
| + | |||
| + | ''UNI'' ''BGUNI'' | ||
| + | ''MESH_SPLIT'' ''MESH_DIM'' ''SZ<sub>1</sub>'' ''SZ<sub>2</sub>'' ... ''SZ<sub>MESH_DIM</sub>'' | ||
| + | ''POINTS_FILE'' | ||
| + | ''FACES_FILE'' | ||
| + | ''OWNER_FILE'' | ||
| + | ''NEIGHBOUR_FILE'' | ||
| + | ''MATERIALS_FILE'' | ||
| + | |||
| + | Defines an unstructured mesh-based geometry. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name for the unstructured mesh-based geometry | ||
| + | |- | ||
| + | | <tt>''BGUNI''</tt> | ||
| + | | : name of the background universe filling all undefined space | ||
| + | |- | ||
| + | | <tt>''MESH_SPLIT''</tt> | ||
| + | | : splitting criterion for the adaptive search mesh (maximum number of geometry cells in search mesh cell) | ||
| + | |- | ||
| + | | <tt>''MESH_DIM''</tt> | ||
| + | | : number of levels in the adaptive search mesh | ||
| + | |- | ||
| + | | <tt>''SZ<sub>i</sub>''</tt> | ||
| + | | : size of the search mesh at level <tt>''i''</tt> | ||
| + | |- | ||
| + | | <tt>''POINTS_FILE''</tt> | ||
| + | | : path to the unstructured mesh points file | ||
| + | |- | ||
| + | | <tt>''FACES_FILE''</tt> | ||
| + | | : path to the unstructured mesh faces file | ||
| + | |- | ||
| + | | <tt>''OWNER_FILE''</tt> | ||
| + | | : path to the unstructured mesh owner file | ||
| + | |- | ||
| + | | <tt>''NEIGHBOUR_FILE''</tt> | ||
| + | | : path to the unstructured mesh neighbour file | ||
| + | |- | ||
| + | | <tt>''MATERIALS_FILE''</tt> | ||
| + | | : path to the unstructured mesh materials file | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *See also description of [[#solid (irregular 3D geometry definition)|solid (irregular 3D geometry definition), type 1]]. | ||
| + | |||
| + | === utrans (universe transformation)<span id="utrans"></span> === | ||
| + | |||
| + | See [[#trans (transformations)|transformations]]. | ||
| + | |||
| + | === wwgen (response matrix based importance map solver)<span id="wwgen"></span> === | ||
| + | |||
| + | '''wwgen''' ''NAME'' ''LIM'' ''NI'' ''MOD'' ''ERG'' ''MSH'' | ||
| + | ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''SZ<sub>1</sub>'' | ||
| + | ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ''SZ<sub>2</sub>'' | ||
| + | ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ''SZ<sub>3</sub>'' | ||
| + | ''DET<sub>1</sub>'' ''W<sub>1</sub>'' [ ''DET<sub>2</sub>'' ''W<sub>2</sub>'' ... ] | ||
| + | |||
| + | '''wwgen''' ''NAME'' ''LIM'' ''NI'' ''MOD'' ''ERG'' ''MSH'' | ||
| + | ''SZ<sub>1</sub>'' ''SZ<sub>2</sub>'' ''SZ<sub>3</sub>'' | ||
| + | ''LIM<sub>11</sub>'' ''LIM<sub>12</sub>'' ... | ||
| + | ''LIM<sub>21</sub>'' ''LIM<sub>22</sub>'' ... | ||
| + | ''LIM<sub>31</sub>'' ''LIM<sub>32</sub>'' ... | ||
| + | ''DET<sub>1</sub>'' ''W<sub>1</sub>'' [ ''DET<sub>2</sub>'' ''W<sub>2</sub>'' ... ] | ||
| + | |||
| + | '''wwgen''' ''NAME'' ''LIM'' ''NI'' ''MOD'' ''ERG'' ''MSH'' | ||
| + | ''X<sub>0</sub>'' ''Y<sub>0</sub>'' | ||
| + | ''P'' ''NX'' ''NY'' | ||
| + | ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ''SZ<sub>3</sub>'' | ||
| + | ''DET<sub>1</sub>'' ''W<sub>1</sub>'' [ ''DET<sub>2</sub>'' ''W<sub>2</sub>'' ... ] | ||
| + | |||
| + | Defines the parameters for importance map calculation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : a unique name to identify the calculation | ||
| + | |- | ||
| + | | <tt>''LIM''</tt> | ||
| + | | : convergence criterion (typical value 1E-12) | ||
| + | |- | ||
| + | | <tt>''NI''</tt> | ||
| + | | : maximum number of iterations | ||
| + | |- | ||
| + | | <tt>''MOD''</tt> | ||
| + | | : solution mode (1 = single detector, 2 = multiple detectors, 3 = global variance reduction) | ||
| + | |- | ||
| + | | <tt>''ERG''</tt> | ||
| + | | : energy group structure (or -1 if no energy dependence is included) | ||
| + | |- | ||
| + | | <tt>''MSH''</tt> | ||
| + | | : mesh type (1 = Cartesian, 2 = Cylindrical, 4 = x-type hexagonal, 5 = y-type hexagonal, 6 = unevenly-spaced xyz, 8 = unevenly spaced cylindrical) | ||
| + | |- | ||
| + | | <tt>''MIN<sub>n</sub>''</tt> | ||
| + | | : minimum mesh boundary (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''MAX<sub>n</sub>''</tt> | ||
| + | | : maximum mesh boundary (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''SZ<sub>n</sub>''</tt> | ||
| + | | : number of mesh cells (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''LIM<sub>nm</sub>''</tt> | ||
| + | | : mesh boundary ''m''th (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>'', ''Y<sub>0</sub>''</tt> | ||
| + | | : mesh center of hexagonal mesh (currently must be centered at the origin) | ||
| + | |- | ||
| + | | <tt>''P''</tt> | ||
| + | | : hexagonal cell pitch | ||
| + | |- | ||
| + | | <tt>''NX'', ''NY''</tt> | ||
| + | | : hexagonal mesh size | ||
| + | |- | ||
| + | | <tt>''DET<sub>i</sub>''</tt> | ||
| + | | : detectors used as target response functions | ||
| + | |- | ||
| + | | <tt>''W<sub>i</sub>''</tt> | ||
| + | | : weight factors for detector scores | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The solution mode provides various options on how the responses are used for calculating the importances. | ||
| + | *The detector entries can be left out in global variance reduction mode (<tt>''MOD''</tt> = 3), in which case the mesh is optimized to uniformly populate the entire geometry. | ||
| + | *Cartesian and cylindrical mesh are defined by outer mesh boundaries and number of mesh cells. | ||
| + | *Unevenly-spaced meshes are defined by providing the mesh cell boundaries separately. | ||
| + | *The coordinate axes 1, 2 and 3 in Cartesian mesh refer to (x,y,z) and in cylindrical mesh to (r,θ,z), with θ given in degrees. | ||
| + | *The hexagonal mesh is defined by mesh center, cell pitch, number of cells in the radial dimensions (similar to the hexagonal lattice) and axial binning. | ||
| + | *The mesh must be defined slightly larger than the geometry (the mesh boundaries should not coincide with the geometry boundaries). | ||
| + | *Source points located on mesh cell boundaries cause fatal errors. | ||
| + | *May not work if source distribution is biased with weight. | ||
| + | *The importance mesh is printed in file <tt>[input].wwd</tt>. | ||
| + | *Importance (weight window) meshes are read using the [[#wwin (weight window mesh definition)|wwin card]]. | ||
| + | *See also practical examples on [[Variance reduction]]. | ||
| + | *This capability is still <u>very much under development</u>. The input syntax may be revised at some point. | ||
| + | |||
| + | === wwin (weight window mesh definition)<span id="wwin"></span> === | ||
| + | |||
| + | '''wwin''' ''NAME'' | ||
| + | [ [[#wwin_wf|'''wf''']] ''FILE'' ''FMT'' ] | ||
| + | [ [[#wwin_wn|'''wn''']] ''F'' ''X'' ''Y'' ''Z'' ''E'' ] | ||
| + | [ [[#wwin_wx|'''wx''']] ''C'' ''G'' ] | ||
| + | [ [[#wwin_wt|'''wt''']] ''SB'' ''TYPE'' ''MIN'' ''MAX'' ] | ||
| + | [ [[#wwin_wi1|'''wi''']] ''ITP'' ''NI'' ''WWG<sub>1</sub>'' ''DF<sub>1</sub>'' ''WWG<sub>2</sub>'' ''DF<sub>2</sub>'' ... ] | ||
| + | [ [[#wwin_wi2|'''wi''']] ''ITP'' ''NI'' ''WWG'' ''NX'' ''NY'' ''NZ'' ''NLOOP'' ''NTRK'' ''ISPL'' ''NSPL'' ''DSPL<sub>1</sub>'' ''SX<sub>1</sub>'' ''SY<sub>1</sub>'' ''SZ<sub>1</sub>'' ''DSPL<sub>2</sub>'' ''SX<sub>2</sub>'' ''SY<sub>2</sub>'' ''SZ<sub>2</sub>'' ...] | ||
| + | |||
| + | Defines a weight window mesh for variance reduction. The first parameter: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : a unique name to identify the mesh | ||
| + | |} | ||
| + | |||
| + | identifies the mesh. The remaining parameters are defined by separate key words followed by the input values. | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Only works in external source simulation mode. | ||
| + | *Importance (weight window) meshes can be generated by running the [[#wwgen (response matrix based importance map solver)|response matrix based solver]], or read in MCNP WWINP format. | ||
| + | *Importance maps can be visualized using the [[#plot (geometry plot definition)|geometry plotter]]. | ||
| + | *See also [[#set wwb|set wwb]] and [[#set maxsplit|set maxsplit]] for setting options for weight windows, splitting and Russian roulette. | ||
| + | *See also practical examples on [[Variance reduction]]. | ||
| + | *This capability is still <u>very much under development</u>. The input syntax may be revised at some point. | ||
| + | |||
| + | |||
| + | Mesh file (<tt>'''wf'''</tt>):<span id="wwin_wf"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : file path and name of the importance mesh file | ||
| + | |- | ||
| + | | <tt>''FMT''</tt> | ||
| + | | : file format (1 = mesh produced by Serpent importance map generator, 2 = MCNP WWINP format weight window mesh file) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *By default the importance map is read from the mesh file and used as-is, the additional options are provided for adjustments. | ||
| + | *Currently the MCNP format only supports simple mesh types (no sub-mesh). | ||
| + | |||
| + | |||
| + | Mesh normalization (<tt>'''wn'''</tt>):<span id="wwin_wn"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''F''</tt> | ||
| + | | : importance for renormalization | ||
| + | |- | ||
| + | | <tt>''X,Y,Z''</tt> | ||
| + | | : coordinates of point used for renormalization | ||
| + | |- | ||
| + | | <tt>''E''</tt> | ||
| + | | : energy used for renormalization | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The importances can be renormalized by fixing the value at a given position and energy. | ||
| + | |||
| + | |||
| + | Mesh adjustment (<tt>'''wx'''</tt>):<span id="wwin_wx"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''C''</tt> | ||
| + | | : constant multiplier for adjusting importances | ||
| + | |- | ||
| + | | <tt>''G''</tt> | ||
| + | | : exponential for adjusting importances | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The importances can be adjusted by constant multiplier <tt>''C''</tt> and exponential factor <tt>''G''</tt> such that <math>F' = CF^G</math>. | ||
| + | |||
| + | |||
| + | Types and options (<tt>'''wt'''</tt>):<span id="wwin_wt"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''SB''</tt> | ||
| + | | : option to set source biasing on (1/yes) or off (0/no) with Serpent-generated importance maps | ||
| + | |- | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : bounds type for Serpent-generated weight-windows (1 = averaged, 2 = segment-wise) | ||
| + | |- | ||
| + | | <tt>''MIN''</tt> | ||
| + | | : minimum truncation limit for importances | ||
| + | |- | ||
| + | | <tt>''MAX''</tt> | ||
| + | | : maximum truncation limit for importances | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Source biasing is currently not available | ||
| + | |||
| + | |||
| + | Weight-window iterations, fixed mesh (<tt>'''wi'''</tt>):<span id="wwin_wi1"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''ITP''</tt> | ||
| + | | : iteration type (1 = fixed mesh) | ||
| + | |- | ||
| + | | <tt>''NI''</tt> | ||
| + | | : number of iterations between Monte Carlo simulation and the response matrix solver | ||
| + | |- | ||
| + | | <tt>''WWG<sub>i</sub>''</tt> | ||
| + | | : name of the WWG-structure used in the iteration | ||
| + | |- | ||
| + | | <tt>''DF<sub>i</sub>''</tt> | ||
| + | | : global density factor | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The fixed mesh option (<tt>''ITP''</tt> = 1) allows performing iterations using a single or multiple meshes generated using the [[#wwgen (response matrix based importance map solver)|response matrix based solver]]. | ||
| + | *The global density factor is a multiplier applied to all material densities. | ||
| + | |||
| + | |||
| + | Weight-window iterations, adaptive mesh (<tt>'''wi'''</tt>):<span id="wwin_wi2"></span> | ||
| + | |||
| + | {| | ||
| + | | <tt>''ITP''</tt> | ||
| + | | : iteration type (2 = geometry-based adaptation, 3 = tracking-based adaptation) | ||
| + | |- | ||
| + | | <tt>''NI''</tt> | ||
| + | | : number of iterations between Monte Carlo simulation and the response matrix solver | ||
| + | |- | ||
| + | | <tt>''WWG''</tt> | ||
| + | | : name of the WWG-structure used in the iteration | ||
| + | |- | ||
| + | | <tt>''NX''</tt> | ||
| + | | : number of x-divisions for the adaptive mesh | ||
| + | |- | ||
| + | | <tt>''NY''</tt> | ||
| + | | : number of y-divisions for the adaptive mesh | ||
| + | |- | ||
| + | | <tt>''NZ''</tt> | ||
| + | | : number of z-divisions for the adaptive mesh | ||
| + | |- | ||
| + | | <tt>''NLOOP''</tt> | ||
| + | | : number of outer iteration loops in generation of adaptive mesh | ||
| + | |- | ||
| + | | <tt>''NTRK''</tt> | ||
| + | | : number of tracks per loop in generation of adaptive mesh | ||
| + | |- | ||
| + | | <tt>''ISPL''</tt> | ||
| + | | : importance split criterion | ||
| + | |- | ||
| + | | <tt>''NSPL''</tt> | ||
| + | | : neighbor split criterion | ||
| + | |- | ||
| + | | <tt>''DSPL<sub>i</sub>''</tt> | ||
| + | | : density split criterion (negative values for mass, positive values for atomic density) | ||
| + | |- | ||
| + | | <tt>''SZ<sub>i</sub>''</tt> | ||
| + | | : minimum cell dimension | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The adaptive mesh option (<tt>''ITP''</tt> = 2 or 3) starts with a coarse base mesh, and refines the resolution iteratively. | ||
| + | *There are two adaptive mesh options. In the geometry-based option (<tt>''ITP''</tt> = 2) Serpent covers the geometry with <tt>''NTRK''</tt> random tracks and splits cells according to density criteria. In the tracking-based option (<tt>''ITP''</tt> = 3) the tracks are started from the source instead. The procedure is repeated <tt>''NLOOP''</tt> times. | ||
| + | *Cell splitting is defined using the <tt>''NX'', ''NY''</tt> and <tt>''NZ''</tt> options. For example <tt>''NX''</tt> = 2, <tt>''NY''</tt> = 2, <tt>''NZ''</tt> = 2 results in each cell being split to 8 sub-cells (octree mesh). For 2D meshes the <tt>''NZ''</tt> parameter must be set to 1. | ||
| + | *Splitting is carried out recursively, until limiting criteria are met. The <tt>''DSPL''</tt> parameters define upper density boundaries and minimum cell sizes for stopping the splits. | ||
| + | *The importance split criterion defines the maximum relative difference between the importances of two adjacent cells. If the criterion is not met, both cells are split. | ||
| + | *The neighbor split criterion defines the maximum number of neighbor allowed for a cell. If the criterion is not met, the cell is split. | ||
| + | |||
| + | == Input options<span id="input options"></span> == | ||
| + | |||
| + | Input options are used to set various calculation parameters that are not included in the main input cards. Each option is identified by key word "set". Optional values are enclosed within square brackets. | ||
| + | |||
| + | === set absrate === | ||
| + | |||
| + | '''set absrate''' ''A'' [ ''MAT'' ] | ||
| + | |||
| + | Sets normalization to total absorption rate. | ||
| + | |||
| + | {| | ||
| + | | <tt>''F''</tt> | ||
| + | | : number of neutrons absorbed per second (neutrons/s) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : dummy parameter | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *Absorption includes all reactions in which the incident neutron is lost, i.e. all capture reactions and fission. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set powdens|set powdens]], [[#set flux|set flux]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set acelib === | ||
| + | |||
| + | '''set acelib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets the cross section directory file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : file paths to directory files | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *If the file path contains special characters it is advised to enclose it within quotes. | ||
| + | *A default directory path can be set by defining environment variable <tt>SERPENT_DATA</tt>. The code looks for cross section directory files in this path if not found at the absolute. | ||
| + | *A default cross section directory file can be set by defining environment variable <tt>SERPENT_ACELIB</tt>. This file will be used if no other path is given with '''set acelib'''. | ||
| + | |||
| + | === set adf === | ||
| + | |||
| + | '''set adf''' ''UNI SURF SYM [ENF]'' | ||
| + | Sets parameters for the calculation of assembly discontinuity factors (ADFs). Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe where spatial homogenization is performed | ||
| + | |- | ||
| + | | <tt>''SURF''</tt> | ||
| + | | : surface enclosing the universe | ||
| + | |- | ||
| + | | <tt>''SYM''</tt> | ||
| + | | : symmetry option ([[ADF symmetry options|see separate list]]) | ||
| + | |- | ||
| + | | <tt>''ENF''</tt> | ||
| + | | : option to skip diffusion flux solver and enforce flat homogeneous flux distribution based on mean heterogeneous flux (1/yes). Default is (0/no). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The surface enclosing the universe can be super-imposed (i.e. not part of the geometry definition), but it must enclose the <u>entire</u> universe. | ||
| + | *The surface is super-imposed on the geometry, i.e. its parameters (coordinates) are relative to the [[Input_syntax_manual#set_root|root universe]] (default 0) | ||
| + | *When the universe is surrounded by zero net-current (reflective) boundary conditions, the ADFs are calculated as the ratios of surface- and volume-averaged heterogeneous flux. | ||
| + | *When the net current is non-zero, the calculation is based on the ratio of surface-averaged homogeneous and heterogeneous flux. The homogeneous flux is obtained from a [[Diffusion flux solver|built-in diffusion flux solver]]. | ||
| + | *Calculation parameters for the diffusion flux solver can be set using the [[#set dfsol|set dfsol]] option. | ||
| + | *Calculation of ADFs is currently allowed only for planes and infinite square and hexagonal prisms. | ||
| + | *Symmetry options are used to average out the statistical variation in the ADFs, which might otherwise lead to systematic errors in core calculations. It is important that the options are used only when the geometry has the corresponding symmetry. | ||
| + | *See separate description of [[Output parameters#Assembly discontinuity factors|output parameters]] in the <tt>[input]_res.m</tt>. | ||
| + | *The ADFs can be printed to the [[Automated_burnup_sequence#Output|group constant output]] with [[Input syntax manual#set_coefpara|set coefpara]]. | ||
| + | *ADFs are calculated in the [[#set nfg|few-group structure used for group constant generation]]. | ||
| + | *The <tt>''ENF''</tt> parameter should be switched on only in rare cases (and you should know what you are doing). | ||
| + | |||
| + | === set alb === | ||
| + | |||
| + | '''set alb''' ''UNI SURF DIR'' | ||
| + | Sets parameters for calculating albedos. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe where spatial homogenization is performed | ||
| + | |- | ||
| + | | <tt>''SURF''</tt> | ||
| + | | : surface for which the albedos are calculated | ||
| + | |- | ||
| + | | <tt>''DIR''</tt> | ||
| + | | : current direction (-1 = inward, 1 = outward) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *When this option is set, Serpent calculates both total albedos (ratio of currents) and partial albedos (response matrix). | ||
| + | *The surface enclosing the universe can be super-imposed (i.e. not part of the geometry definition), but it must enclose the <u>entire</u> universe. | ||
| + | *The surface is super-imposed on the geometry, i.e. its parameters (coordinates) are relative to the [[Input_syntax_manual#set_root|root universe]] (default 0) | ||
| + | *The current direction is given relative to the surface normal vectors | ||
| + | *The universe is needed only for labelling the results in the output files. | ||
| + | *See separate description of [[Output parameters#Albedos|output parameters]]. | ||
| + | *Albedos are calculated in the [[#set nfg|few-group structure used for group constant generation]]. | ||
| + | |||
| + | === set arr === | ||
| + | |||
| + | '''set arr''' ''MODEN'' [ ''MODEG'' ] | ||
| + | Sets analog reaction rate calculation on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODEN''</tt> | ||
| + | | : mode for neutrons (0 = no reactions included, 1 = include only reactions that affect neutron balance, 2 = include all reactions) | ||
| + | |- | ||
| + | | <tt>''MODEG''</tt> | ||
| + | | : mode for photons (0 = no reactions included, 1 = include all reactions) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Analog reaction rates are calculated by counting sampled events and printed in a separate output file <tt>[input]_arr[bu].m</tt>, where "<tt>bu</tt>" is the burnup step. | ||
| + | *See detailed description on the [[Description of output files#Reaction rate output|reaction rate output file]]. | ||
| + | |||
| + | === set ba === | ||
| + | |||
| + | '''set ba''' ''ZAI<sub>1</sub> ZAI<sub>2</sub> ...'' | ||
| + | Defines isotopes handled separately as burnable absorbers. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''ZAI<sub>n</sub>''</tt> | ||
| + | | : nuclide identifiers ([[Definitions, units and constants#definitions|ZAI]]) | ||
| + | |} | ||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Some burnup applications require separate treatment for isotopes that are used as burnable absorbers but also produced in fission. This input parameter can be used to separate the transmutation chains. | ||
| + | *Isotope handled as the burnable absorber is created by duplicating the original and renaming it as ''ZAI<sub>n</sub> + 1000''. | ||
| + | *For Gd-155, for example, the fission product isotope would be assigned ZAI 641550 and the burnable absorber ZAI 642550. | ||
| + | *The input parameter defines the entire transmutation chain. Listing Gd-isotopes 641540 641550 641560 641570 641580 creates a transmutation path from Gd-154 to Gd-158. Listing only the main absorbers (641550 641570) produces a different result, since the capture products of Gd-155 and Gd-157 are lost. | ||
| + | |||
| + | === set bala === | ||
| + | |||
| + | '''set bala''' ''OPT'' | ||
| + | Sets OpenMP load balancing on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : probability to store particles in common queue (0 = off, >0 = on) | ||
| + | |} | ||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Load balancing may improve OpenMP parallel scalability in calculations with significant branching (most typically related to coupled neutron/photon calculations or variance reduction). | ||
| + | *The option is ON with <tt>''OPT''</tt>= 1 with weight-window/variance reduction calculations and dynamic/time-dependent calculation modes. Otherwise, it is set OFF by default. Before version 2.2.0, the default behavior was always OFF. | ||
| + | *When this option is set, the random number sequence is no longer preserved. | ||
| + | |||
| + | === set bc === | ||
| + | |||
| + | '''set bc''' ''MODE'' | ||
| + | Sets the boundary conditions for all outer boundaries of the geometry. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : boundary type (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |} | ||
| + | |||
| + | '''set bc''' ''MODE ALB'' | ||
| + | Sets the boundary conditions with albedo for all outer boundaries of the geometry. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : boundary type (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''ALB''</tt> | ||
| + | | : albedo | ||
| + | |} | ||
| + | |||
| + | '''set bc''' ''MODEX MODEY MODEZ'' | ||
| + | Sets the boundary conditions separately for x-, y- and z-directions. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODEX''</tt> | ||
| + | | : boundary type in x-direction (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''MODEY''</tt> | ||
| + | | : boundary type in y-direction (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''MODEZ''</tt> | ||
| + | | : boundary type in z-direction (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | '''set bc''' ''MODEX MODEY MODEZ ALB'' | ||
| + | Sets the boundary conditions with albedo separately for x-, y- and z-directions. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODEX''</tt> | ||
| + | | : boundary type in x-direction (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''MODEY''</tt> | ||
| + | | : boundary type in y-direction (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''MODEZ''</tt> | ||
| + | | : boundary type in z-direction (1 = vacuum, 2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''ALB''</tt> | ||
| + | | : albedo | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The boundary conditions can be set either for all directions at once (single parameter) or x-, y- and z-directions separately (three parameters). Albedos are provided by adding one more parameter in the list. | ||
| + | *The default boundary condition is vacuum (= 1) in all directions. | ||
| + | *Albedo boundary conditions are invoked by multiplying the particle weight with factor <tt>''ALB''</tt> each time a reflective or periodic boundary is hit. | ||
| + | *Repeated boundary conditions (reflective or periodic) are based on universe transformations, which limits outer boundary to surfaces that form regular lattices (square and hexagonal prisms, rectangles, cubes and cuboids). | ||
| + | *Repeated boundary conditions are applied on the first surface of outside cells (see definition of outside cells in the [[#cell (cell definition)|cell card]]) | ||
| + | *For symmetry purposes Serpent provides the [[#set usym|universe symmetry option]]. | ||
| + | *For more information, see [[Boundary conditions|detailed description on boundary conditions]]. | ||
| + | *The boundary condition type numbers can also be given as strings, with black = 1, reflective = 2 and periodic = 3. | ||
| + | |||
| + | === set blockdt === | ||
| + | |||
| + | '''set blockdt''' ''MAT<sub>1</sub> MAT<sub>2</sub> ...'' | ||
| + | Defines the list of materials where delta-tracking is never used. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>n</sub>''</tt> | ||
| + | | : material names | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *This option is used to override selection of tracking mode based on the probability threshold (see [[#set dt|set dt]]) in individual materials. | ||
| + | *Use of delta-tracking can be forced in individual materials using [[#set forcedt|set forcedt]]. | ||
| + | *For more information on tracking modes, see the detailed description on [[delta- and surface-tracking]]. | ||
| + | *''Note to developers: should have different lists for neutrons and photons?'' | ||
| + | |||
| + | === set bralib === | ||
| + | |||
| + | '''set bralib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets isomeric branching data library file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : library file paths | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Isomeric branching data libraries are standard ENDF format files containing energy-dependent branching ratios. The data is read from ENDF files 9 and 10. | ||
| + | *Serpent uses [[Default isomeric branching ratios|constant branching ratios]] by default. The default values can be overridden using the [[#set isobra|set isobra]] option. Energy-dependent data read read from ENDF format files overrides the constant ratios. | ||
| + | *If the file path contains special characters it is advised to enclose it within quotes. | ||
| + | *A default directory path can be set by defining environment variable SERPENT_DATA. The code looks for decay data files in this path if not found at the absolute. | ||
| + | *See also: [[Branching ratio example|example input]] | ||
| + | *Example data from the [http://virtual.vtt.fi/virtual/montecarlo/misc/sss_jeff31a.bra JEFF-3.1 activation file] | ||
| + | |||
| + | === set branchless === | ||
| + | |||
| + | '''set branchless''' ''OPT'' [ ''WGT_LOW'' ''WGT_HIGH'' ] | ||
| + | Option that enables the branchless collision method for variance reduction. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch calculation on (1/yes) or off (0/no). Default is off. | ||
| + | |- | ||
| + | | <tt>''WGT_LOW''</tt> | ||
| + | | : weight lower-boundary (default value: 0.2) | ||
| + | |- | ||
| + | | <tt>''WGT_HIGH''</tt> | ||
| + | | : weight upper-boundary (default value: 10.0) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The branchless algorithm suppresses the variability due to the simultaneous propagation of the several branches associated to a fission event | ||
| + | *The branchless method uses analog scattering combined with forced fission so that after each collision, the neutron is either a scattering neutron or a fission neutron. In a non-multiplying method, the branchless method behaves as implicit capture. | ||
| + | |||
| + | === set bumode === | ||
| + | |||
| + | '''set bumode''' ''MODE'' [ ''ORDER'' ''SSD'' ] | ||
| + | |||
| + | Sets the burnup calculation mode. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : burnup calculation mode | ||
| + | |- | ||
| + | | <tt>''ORDER''</tt> | ||
| + | | : CRAM order | ||
| + | |- | ||
| + | | <tt>''SSD''</tt> | ||
| + | | : number of substeps for CRAM decay steps (default or 0: use TTA) | ||
| + | |} | ||
| + | |||
| + | The possible settings for mode are: | ||
| + | |||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Mode | ||
| + | ! Description | ||
| + | |- | ||
| + | | <tt>1, tta</tt> | ||
| + | | Transmutation Trajectory Analysis (TTA) | ||
| + | |- | ||
| + | | <tt>2, cram</tt> | ||
| + | | Chebyshev Rational Approximation Method (CRAM) | ||
| + | |} | ||
| + | |||
| + | The CRAM order parameter can only be given when choosing the CRAM mode. The possible settings for CRAM order are: | ||
| + | |||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! CRAM order | ||
| + | |- | ||
| + | | <tt>2</tt> | ||
| + | |- | ||
| + | | <tt>4</tt> | ||
| + | |- | ||
| + | | <tt>6</tt> | ||
| + | |- | ||
| + | | <tt>8</tt> | ||
| + | |- | ||
| + | | <tt>10</tt> | ||
| + | |- | ||
| + | | <tt>12</tt> | ||
| + | |- | ||
| + | | <tt>14</tt> | ||
| + | |- | ||
| + | | <tt>16</tt> | ||
| + | |- | ||
| + | | <tt>-16</tt> | ||
| + | |- | ||
| + | | <tt>-48</tt> | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default setting for the burnup calculation mode is CRAM. | ||
| + | *Default value for the CRAM order is 14 resulting in order 14 PFD CRAM. | ||
| + | *Negative values of CRAM order mean using IPF form of CRAM with order of the absolute value of the parameter. | ||
| + | *Positive values refer to PFD form of CRAM. | ||
| + | *Decay calculations (see [[#dep (depletion history)|dep (depletion history)]]) and burnup calculations with very low flux are always calculated with TTA disregarding this input before version 2.1.32. The latter, very low flux condition, only applies to calculations not involving continuous reprocessing. | ||
| + | *Positive values of <tt>''SSD''</tt> enforce usage of CRAM with given number of substeps. | ||
| + | *The Serpent 1 <tt>''MODE''</tt> 3, a variation TTA method, in which cyclic transmutation chains are handled by inducing small variations in the coefficients instead of solving the extended TTA equations, is overwritten by the standard TTA method <tt>''MODE''</tt> 1. | ||
| + | *Version 2.2.0 includes the sub-step method for depletion calculations involving continuous reprocessing. | ||
| + | |||
| + | === set bunorm === | ||
| + | |||
| + | '''set bunorm''' ''NORM'' | ||
| + | |||
| + | Sets the burnup calculation normalization mode if it is not bound to a single material. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NORM''</tt> | ||
| + | | : burnup calculation normalization mode (1 = all materials, 2 = burnable materials, 3 = non-burnable materials) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * The default normalization for burnup calculations includes all materials. | ||
| + | |||
| + | === set ccmaxiter === | ||
| + | |||
| + | '''set ccmaxiter''' ''NITER'' | ||
| + | Sets the maximum number of coupled calculation iterations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NITER''</tt> | ||
| + | | : number of iterations. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Default maximum number of iterations is 1 (no iteration). | ||
| + | *The iteration is stopped when either the maximum number of iterations or the maximum active neutron population (set with [[#set ccmaxpop|set ccmaxpop]]) has been simulated. | ||
| + | *See [[Coupled multi-physics calculations]] for further information. | ||
| + | |||
| + | === set ccmaxpop === | ||
| + | |||
| + | '''set ccmaxpop''' ''CPOP'' | ||
| + | Sets the maximum total live population to simulate in a coupled calculation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''CPOP''</tt> | ||
| + | | : total active population to simulate. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Default maximum population is [[Definitions, units and constants#Constants|INFTY]]/1e6. | ||
| + | *The iteration is stopped when either the maximum number of iterations (set with [[#set ccmaxiter|set ccmaxiter]]) or the maximum active neutron population has been simulated. | ||
| + | *Only the population simulated during active cycles is included in this amount. | ||
| + | *This is mostly useful if the neutron population per iteration is not constant. | ||
| + | *See [[Coupled multi-physics calculations]] for further information. | ||
| + | |||
| + | === set cdop === | ||
| + | |||
| + | '''set cdop''' ''OPT'' | ||
| + | Sets the Doppler broadening method for the energy spectrum of the scattered photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set Doppler broadening method off (0/no) or on (1/yes). The default option is on. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *If the Doppler broadening method is switched off, the incoherent scattering function approximation is used for calculating the energy. | ||
| + | *In both cases, the direction of the photon is calculated using the incoherent scattering function. | ||
| + | |||
| + | === set cea === | ||
| + | |||
| + | '''set cea''' ''OPT'' | ||
| + | Sets the Compton electron angular distribution model on and off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set the Compton electron angular distribution model off (0/no) or on (1/yes). The default option is on. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Electron travels in the direction of the momentum transfer vector. This is equal to the free-electron scattering angle when Doppler broadening is not used. | ||
| + | |||
| + | === set cfe === | ||
| + | |||
| + | '''set cfe''' ''LN'' [ ''TN LG TG'' ] | ||
| + | Defines the minimum mean distance for scoring the collision flux estimator (CFE) for photons and neutrons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LN''</tt> | ||
| + | | : minimum mean distance for scoring the CFE for neutrons | ||
| + | |- | ||
| + | | <tt>''TN''</tt> | ||
| + | | : minimum mean time interval for scoring the CFE for neutrons | ||
| + | |- | ||
| + | | <tt>''LG''</tt> | ||
| + | | : minimum mean distance for scoring the CFE for photons | ||
| + | |- | ||
| + | | <tt>''TG''</tt> | ||
| + | | : minimum mean time interval for scoring the CFE for photons | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The use of delta-tracking necessitates the use of CFE for scoring the integral reaction rates. The scoring is based on both real and virtual collision to improve the statistics in low density regions (and short time intervals). | ||
| + | *The minimum mean distance is the statistical mean-free-path (mfp) of collisions that contribute to the CFE. Collisions are more frequent if the physical mfp is shorter. | ||
| + | *In time-dependent simulations it may be more convenient to define the minimum mean time between two collisions, to get sufficient statistics for short time bins. | ||
| + | *The default minimum mean scoring distance is 20 cm for both neutrons and photons. Adjusting the distance affects both statistics and running time, but it should be noted that <u>no studies have been performed on what the optimal value should be</u>. | ||
| + | *Only one criterion can be provided for each particle type. If distance is given, time must be set to -1 and vice versa. | ||
| + | *For more information on tracking modes and CFE, see the detailed descriptions on [[delta- and surface-tracking]] and [[Result estimators#Implicit estimators|result estimators]]. | ||
| + | *The collision flux estimator in Serpent is described in an article in Annals of Nuclear energy from 2017.<ref name="cfe">Leppänen, J. | ||
| + | ''"On the use of delta-tracking and the collision flux estimator in the Serpent 2 Monte Carlo particle transport code."'' Ann. Nucl. Energy [http://www.sciencedirect.com/science/article/pii/S0306454916311367 '''105''' (2017) 161-167].</ref> | ||
| + | *In version 2.1.27 and earlier the name of this input option was "set minxs". | ||
| + | |||
| + | === set cmm === | ||
| + | |||
| + | '''set cmm''' ''OPT'' | ||
| + | Sets calculation of diffusion coefficients using the cumulative migration method (CMM) on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch CMM calculation on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Calculation of [[Output_parameters#Diffusion_parameters|diffusion coefficients using CMM]] might take considerable time. This option allows switching the calculation off if the data is not needed. | ||
| + | *The calculation of CMM diffusion coefficients was revised in version 2.1.31 so that the calculated values may be different than with previous versions. | ||
| + | *CMM diffusion coefficients can be calculated also when using [[#set_impl|implicit capture reactions]] beginning from version 2.1.31. | ||
| + | *CMM diffusion coefficients and transport cross sections are reasonable only when they are calculated over entire geometry (homogenized region covers the entire geometry and is surrounded by periodic or reflective boundary conditions). | ||
| + | **This means that e.g. pin cell diffusion coefficients can not be calculated from a 2D fuel assembly calculation. | ||
| + | **One may try to approximate the CMM diffusion coefficients with [[#set_trc|TRC diffusion coefficients]] with transport correction for hydrogen for light water reactor applications. | ||
| + | *Using [[#set_shbuf|private results array]] may be recommended when CMM diffusion coefficients are calculated. | ||
| + | |||
| + | === set coefpara === | ||
| + | |||
| + | '''set coefpara''' ''FMT'' [ ''PARAM<sub>1</sub> PARAM<sub>2</sub>'' ... ] | ||
| + | Defines the parameters included in the separate group constant output file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FMT''</tt> | ||
| + | | : output format, currently used for including or excluding statistical errors (0 = not included, 1 = included) | ||
| + | |- | ||
| + | | <tt>''PARAM<sub>n</sub>''</tt> | ||
| + | | : list of parameters or detectors included in the file | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The group constant output file <tt>[input].coe</tt> is produced when the [[automated burnup sequence]] is invoked. | ||
| + | *The available parameters are listed under [[Output parameters#Homogenized group constants|homogenized group constants]] in the description of the <tt>[input]_res.m</tt> output file. | ||
| + | *Detectors are identified by the name assigned to them in the [[#det (detector definition)|detector card]]. | ||
| + | |||
| + | === set combing === | ||
| + | |||
| + | '''set combing''' ''MODE'' | ||
| + | Option that enables the combing approach for precursors population control as an alternative to Russian roulette and splitting in dynamic source simulations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : combing population-control mode (0 = none, 1 = weight-based, 2 = emission-based) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The combing method can achieve variance reduction and save computer time by keeping the population size approximately constant over time steps. In super-critical systems, it prevents the population from growing without bound while, in sub-critical systems, it does it from dying. In critical systems, it avoids the divergence of the variance of the population due to fluctuations of fission chains. | ||
| + | |||
| + | === set comfile === | ||
| + | |||
| + | '''set comfile''' ''INFILE'' ''OUTFILE'' | ||
| + | Defines the communication files used in the file-based coupled calculation communications. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''INFILE''</tt> | ||
| + | | : Path to inwards communication file (signals to Serpent). | ||
| + | |- | ||
| + | | <tt>''OUTFILE''</tt> | ||
| + | | : Path to outwards communication file (signals from Serpent). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Setting up a communication mode will enable the coupled calculation mode. | ||
| + | *The communication options [[#set comfile|set comfile]], [[#set ppid|set ppid]] and [[#set pport|set pport]] are mutually exclusive, aka, multiple signalling modes are not allowed. | ||
| + | *For more information see: [[Coupled_multi-physics_calculations#External_coupling|External coupling]] | ||
| + | |||
| + | === set confi === | ||
| + | |||
| + | '''set confi''' ''OPT'' | ||
| + | Sets confidentiality flag on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set confidentiality flag on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *This option can be used to label calculations as confidential. If the option is set, text "(CONFIDENTIAL)" is printed in the run-time output next to the [[#set title|calculation title]] and the value of variable CONFIDENTIAL_DATA in the <tt>[input]_res.m</tt> output file is set to 1. | ||
| + | |||
| + | === set coverxlib === | ||
| + | |||
| + | '''set coverxlib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets COVERX-format multi-group covariance data file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : file paths to multi-group covariance data files in the COVERX format (ASCII or binary) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *If covariance data is linked when running [[Sensitivity calculations]], Serpent will automatically apply the sandwich rule using the calculated sensitivity vectors and propagate the covariance data to uncertainties of the sensitivity responses. | ||
| + | |||
| + | === set covlib === | ||
| + | |||
| + | '''set covlib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets plain ASCII multi-group covariance data file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : file paths to multi-group covariance data files in the plain ASCII format (ASCII or binary) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *If covariance data is linked when running [[Sensitivity calculations]], Serpent will automatically apply the sandwich rule using the calculated sensitivity vectors and propagate the covariance data to uncertainties of the sensitivity responses. | ||
| + | |||
| + | === set cpd === | ||
| + | |||
| + | '''set cpd''' ''DEPTH'' [ ''N<sub>Z</sub> Z<sub>MIN</sub> Z<sub>MAX</sub>'' ] [ ''LVL1'' ''LVL2'' ] | ||
| + | Sets on the calculation of lattice-wise power distributions to output file <tt>[input]_core0.m</tt> on. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''DEPTH''</tt> | ||
| + | | : The number of levels included. 1 is the first lattice calculated from universe 0 usually corresponding to assembly-wise distribution. 2 includes the first two levels usually corresponding to the assembly- and pin-wise distributions. | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : Number of equal sized axial bins into which the lattices are divided. | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : Minimum z-coordinate for the axial division. | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : Maximum z-coordinate for the axial division. | ||
| + | |- | ||
| + | | <tt>''LVL1''</tt> | ||
| + | |: User-defined first level where to define the lattice-wise power distribution | ||
| + | |- | ||
| + | | <tt>''LVL2''</tt> | ||
| + | |: User-defined second level where to define the lattice-wise power distribution | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * The default values for <tt>''N<sub>Z</sub>''</tt>, <tt>''Z<sub>MIN</sub>''</tt> and <tt>''Z<sub>MAX</sub>''</tt> are 1, -INFTY and INFTY, respectively. | ||
| + | |||
| + | === set cpop === | ||
| + | |||
| + | '''set cpop''' ''NPG'' ''NGEN'' ''NSKIP'' [ ''NSKIP2'' ] | ||
| + | Sets parameters for simulated neutron population for corrector neutron transport solutions in burnup calculation. Typically used with the [[Stochastic Implicit Euler burnup scheme|SIE burnup scheme]]. Input values | ||
| + | |||
| + | {| | ||
| + | | <tt>''NPG''</tt> | ||
| + | | : number of neutrons per generation | ||
| + | |- | ||
| + | | <tt>''NGEN''</tt> | ||
| + | | : number of active generations | ||
| + | |- | ||
| + | | <tt>''NSKIP''</tt> | ||
| + | | : number of inactive generations | ||
| + | |- | ||
| + | | <tt>''NSKIP2''</tt> | ||
| + | | : number of inactive generations on further iterations for the same burnup point | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *As the SIE burnup scheme executes the corrector step multiple times for each burnup step, combining the results from each iteration, it may be a good idea to run more iterations with less active neutron histories per iteration. | ||
| + | |||
| + | === set csw === | ||
| + | |||
| + | '''set csw''' ''FILE'' | ||
| + | Writes source points in criticality source simulation into a file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : file name where the source points are written | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Only source points from active cycles are included. | ||
| + | |||
| + | === set dataout === | ||
| + | |||
| + | '''set dataout''' ''TABLE_LIST'' | ||
| + | Defines the tables included in the nuclear and material data file <tt>[input].out.m</tt>. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TABLE_LIST''</tt> | ||
| + | | : list of tables | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The file lists all nuclides and their reactions as they are read from the nuclear data libraries. The material data includes isotopic compositions and densities, as well as volumes and masses if available. (See the [[Description of output files#Nuclide and material data output|description of the nuclear and material data output]]). | ||
| + | *The data is divided into two sections: nuclear data (Tables 1-4) and material data (Table 5), and the tables are the following: | ||
| + | **Table 1: Summary of nuclide data | ||
| + | **Table 2: Reaction and decay data | ||
| + | **Table 3: Fission yield data (only in burnup mode) | ||
| + | **Table 4: Lost transmutation paths (only in burnup mode) | ||
| + | **Table 5: Summary of material compositions | ||
| + | *The entry-names to input the tables are: "all"/0 = Tables 1-5, "nuc_summary"/1 = Tables 1, "nuc_readec"/2 = Table 2, "nuc_nfy"/3 = Table 3, "nuc_lostpath"/4 = Table 4, "mat_summary"/5 = Table 5, "allnuc"/8 = Tables 1-4, "allmat"/9 = Table 5. | ||
| + | *The default behavior is to include all the tables/data available. To omit the <tt>[input].out</tt> file, select "-1". | ||
| + | |||
| + | === set dbrc === | ||
| + | |||
| + | '''set dbrc''' ''E<sub>min</sub> E<sub>max</sub> NUC<sub>1</sub>'' [ ''NUC<sub>2</sub>'' ... ] | ||
| + | Enables the use of doppler-broadening rejection correction (DBRC). Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''E<sub>min</sub>''</tt> | ||
| + | | : Minimum energy for DBRC | ||
| + | |- | ||
| + | | <tt>''E<sub>max</sub>''</tt> | ||
| + | | : Maximum energy for DBRC | ||
| + | |- | ||
| + | | <tt>''NUC<sub>n</sub>''</tt> | ||
| + | | : Nuclide identifiers for which to apply DBRC to, with 0 K cross section data, e.g. "92238.00c" | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This description is not complete. | ||
| + | *Use of DBRC requires 0 K cross section data. | ||
| + | *See also Section 5.6 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | *This input could be given without any nuclides before version 2.1.32. Then DBRC was not used at all. | ||
| + | |||
| + | === set dd === | ||
| + | |||
| + | '''set dd''' ''MODE'' [ ''X<sub>0</sub>'' ''Y<sub>0</sub>'' ''α<sub>0</sub>'' ] | ||
| + | Invokes domain decomposition. Input values | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : decomposition mode (0 = none, 1 = simple, 2 = sector, 3 = sector + center) | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | x-coordinate of the domain decomposition origin (origin is the centre of the angular division) | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | y-coordinate of the domain decomposition origin (origin is the centre of the angular division) | ||
| + | |- | ||
| + | | <tt>''α<sub>0</sub>''</tt> | ||
| + | | angular position of the domain decomposition origin (origin is the initial position of the angular division) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Domain decomposition works in MPI mode by separating burnable materials into different parallel tasks. | ||
| + | *Number of domains is given by the number of MPI tasks | ||
| + | *Only burnable materials separated into depletion zones using the "div sep" option are decomposed | ||
| + | *<tt>''MODE''</tt> 1 decomposes the geometry based on the automatically assigned depletion zone indexes (not recommended). | ||
| + | *<tt>''MODE''</tt> 2 decomposes the zones into sectors and <tt>''MODE''</tt> 3 adds a central zone if the number of domains is greater than 4. | ||
| + | *The additional input options, <tt>''X<sub>0</sub>''</tt>, <tt>''Y<sub>0</sub>''</tt> and <tt>''α<sub>0</sub>''</tt>, are available for <tt>''MODE''</tt> 2 and 3 (default values are zero). | ||
| + | *Decomposed materials are plotted in domain-specific colors (unless the '''rgb''' entry in the [[#mat (material definition)|material card]] is used) | ||
| + | *See [[Domain decomposition|practical example]] for more information. | ||
| + | |||
| + | === set declib === | ||
| + | |||
| + | '''set declib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets the decay data library file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : library file paths | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Decay libraries are standard ENDF format files containing decay data. | ||
| + | *If the file path contains special characters it is advised to enclose it within quotes. | ||
| + | *A default directory path can be set by defining environment variable <tt>SERPENT_DATA</tt>. The code looks for decay data files in this path if not found at the absolute. | ||
| + | *From version 2.2.0 and on, a default decay data library directory file can be set by defining environment variable <tt>SERPENT_DECLIB</tt>. This file will be used if no other path is given with '''set declib'''. | ||
| + | |||
| + | === set decomp === | ||
| + | |||
| + | '''set decomp''' ''OPT'' [ ''ELEM<sub>1</sub>'' ''ELEM<sub>2</sub>'' ... ] | ||
| + | Decomposes elemental entries in material cards into isotopes. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to include (1) or exclude (0) elements from decomposed list | ||
| + | |- | ||
| + | | <tt>''ELEM<sub>n</sub>''</tt> | ||
| + | | : element names | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| − | + | * Elemental entries are identifed from zero A in ZA. | |
| + | * The decomposition is based on built-in isotope fractions. | ||
| + | * If the list is not provided, all elemental entries are decomposed. | ||
=== set delnu === | === set delnu === | ||
| − | + | '''set delnu''' ''OPT'' | |
Sets delayed neutron emission on or off. Input values: | Sets delayed neutron emission on or off. Input values: | ||
{| | {| | ||
| − | | < | + | | <tt>''OPT''</tt> |
| − | | option to switch delayed neutron emission | + | | : option to switch delayed neutron emission on (1/yes) or off (0/no) |
|} | |} | ||
| − | < | + | <u>Notes:</u> |
| − | *Delayed neutron emission is on by default in neutron criticality source and off by default in external source simulations. | + | *Delayed neutron emission is on by default in neutron criticality source and off by default in (static/dynamic) external source simulation mode. |
| + | *In time-dependent calculations, driven by the [[#set dynsrc|set dynsrc]] option, precursor based delayed neutron emission is included in the calculation: off at fission, but on at delayed nubar in total nubar. | ||
| + | *See separate description of [[physics options in Serpent]] for differences to other codes. | ||
| + | |||
| + | === set depmtx === | ||
| + | '''set depmtx''' ''MODE'' | ||
| + | |||
| + | Print burnup matrixes to <tt>[input]_depmtx_[mat]_[bu]_[ss].m</tt> file during burnup calculation, where "<tt>bu</tt>" is the burnup step and "<tt>ss</tt>" is the substep. | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : Set printing on (1/yes) or off (0/no). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *With non-constant predictor, this option will stop the simulation up to version 2.1.31. | ||
| + | *With multiple substeps, only the last one is kept after the simulation up to version 2.1.31. | ||
| + | *The burnup matrix output is named <tt>depmtx_[mat][bu].m</tt> up to version 2.1.31. | ||
| + | |||
| + | === set depout === | ||
| + | '''set depout''' ''MODE'' [''STEP''] | ||
| + | Controls which burnable material compositions are printed into the <tt>[input]_dep.m</tt> output file in case of divided materials. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : value indicating, which materials to output to the <tt>[input]_dep.m</tt> file (1 = only partials, 2 = only parents, 3 = both) | ||
| + | |- | ||
| + | |<tt>''STEP''</tt> | ||
| + | | : value indicating the print-out interval of the <tt>[input]_dep.m</tt> file (0 = final step, 1 = all steps, 2 =none) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Parent materials refer to materials defined by [[#mat (material definition)|mat cards]], and partials to depletion zones created automatically using the [[#div (divisor definition)|div card]]. | ||
| + | *Default mode is 2 (only parents) and default print-out interval step is 1 (all steps). | ||
| + | *Print-out interval step option 2, no <tt>[input]_dep.m</tt> generation, can be combined with post-processing re-depletion: "-rdep" command line option. | ||
| + | *This option when is used with the domain decomposition feature, [[#set dd|set dd]], in a mode different from 2, generates multiple depletion files which are named adding <tt>_dd[mpiid]</tt> (domain decomposition identifier) to the standard file name. Each of them contains the partial materials information of the given domain/MPI task. | ||
| + | |||
| + | === set deppara === | ||
| + | |||
| + | '''set deppara''' ''PARAM_LIST'' | ||
| + | Defines the variables included in the depletion output file <tt>[input]_dep.m</tt>. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''PARAM_LIST''</tt> | ||
| + | | : list of variables | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default behavior is to include the full-set of variables: atomic density, mass density, activity, decay heat, spontaneous fission rate, photon emission rate, ingestion toxicity, and inhalation toxicity (see the [[Description of output files#Burnup calculation output|description of the depletion output]]). | ||
| + | *The entry-names to input the variables are: "atom" = atomic density, "mass" = mass density, "activity" = activity, "dh" = decay heat, "sf" = spontaneous fission rate, "gsrc" = photon emission rate, "ingtox" = ingestion toxicity, "inhtox" = inhalation toxicity. | ||
| + | *Alternatively, the "all" entry would include the full-set of variables, and the "none" entry would exclude the full-set of variables. | ||
| + | |||
| + | === set depstepbunorm === | ||
| + | |||
| + | '''set depstepbunorm''' ''NORM'' | ||
| + | |||
| + | Sets the depletion step normalization in burnup calculations based on energy deposition. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NORM''</tt> | ||
| + | | depletion step normalization mode based on energy deposition (1 = all materials, 2 = burnable materials) | ||
| + | |} | ||
| + | |||
| + | <u>Notes</u> | ||
| + | * By default, for energy deposition modes 0/1, the normalization includes only "burnable" materials while for energy deposition modes 2/3, the normalization includes "all" materials (see [[#set edepmode|set edepmode]]). | ||
| + | |||
| + | === set dfsol === | ||
| + | '''set dfsol''' ''MODE'' [ ''DC'' ''NP'' ] | ||
| + | Options for homogeneous diffusion flux solver. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : boundary conditions for solver (1 = include net currents at boundary surfaces and corners, 2 = include only surface currents) | ||
| + | |- | ||
| + | | <tt>''DC''</tt> | ||
| + | | : type of diffusion coefficient used in the calculation (1 = INF_DIFFCOEF, 2 = TRC_DIFFCOEF) | ||
| + | |- | ||
| + | | <tt>''NP''</tt> | ||
| + | | : number of points for trapezoidal integration for homogeneous flux | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This input option is used to control how the deterministic diffusion flux solver used to obtain assembly discontinuity factors [[#set adf|(set adf)]] and pin power distributions [[#set ppw|(set ppw)]] is run. | ||
| + | *Default mode is 1 (include both surfaces and corners in solution). | ||
| + | *Default diffusion coefficient is INF_DIFFCOEF, option 2 requires the [[#set trc|set trc]] option. | ||
| + | *Default number of points for trapezoidal integration is 100. | ||
| + | *See also separate description of the [[Diffusion flux solver|built-in diffusion flux solver]]. | ||
| + | *The format was revised in update 2.1.27 (<tt>''DC''</tt> option was added between <tt>''MODE''</tt> and <tt>''NP''</tt>). | ||
| + | |||
| + | === set dix === | ||
| + | '''set dix''' ''OPT'' | ||
| + | Sets double indexing for cross section energy grid look-up on or off: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : option to set double indexing on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Double indexing<ref name="dix">Leppänen, J. | ||
| + | ''"Two practical methods for unionized energy grid construction in continuous-energy Monte Carlo neutron transport calculation."'' Ann. Nucl. Energy [https://www.sciencedirect.com/science/article/pii/S0306454909001108 '''36''' (2009) 878-885].</ref> is a method to speed-up the cross section look-up when energy grid unionization is not used for microscopic data. | ||
| + | *The method can be used only in [[#set_opti|optimization modes]] 1 and 3 (modes 2 and 4 are based on energy grid unionization). | ||
| + | |||
| + | === set dspec === | ||
| + | |||
| + | '''set dspec''' ''EGRID<sub>p</sub>'' ''EGRID<sub>n</sub>'' | ||
| + | Sets the energy grid structure for decay spectra. | ||
| + | |||
| + | {| | ||
| + | | <tt>''EGRID<sub>p</sub>''</tt> | ||
| + | | : energy grid structure for photons | ||
| + | |- | ||
| + | | <tt>''EGRID<sub>n</sub>''</tt> | ||
| + | | : energy grid structure for neutrons | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The photon/neutron decay spectra is printed in the <tt>[input]_gsrc.m</tt> or <tt>[input]_nsrc.m</tt> output file, respectively. | ||
| + | *The energy group spectra only include the contribution from the discrete/line spectra. | ||
| + | *Setting "-1" instead of providing the energy grid structure disables the option for the given particle type. | ||
| + | |||
| + | === set dt === | ||
| + | |||
| + | '''set dt''' ''NTRSH'' [ ''GTRSH'' ] | ||
| + | Sets probability threshold for delta-tracking. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NTRSH''</tt> | ||
| + | | : probability threshold for neutrons | ||
| + | |- | ||
| + | | <tt>''GTRSH''</tt> | ||
| + | | : probability threshold for photons | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Serpent uses delta-tracking by default for both neutrons and photons, but switches to surface-tracking if the probability of sampling virtual collisions (ratio between material total cross section and the majorant) exceeds the given threshold. | ||
| + | *Default probability threshold for both particle types is 0.9, i.e. delta-tracking is used if the ratio between total cross section and majorant is above 0.1. | ||
| + | *"set dt 1" means that delta-tracking is always used and "set dt 0" that it is never used. | ||
| + | *Use of delta-tracking can be enforced or blocked in individual materials using the [[#set forcedt|set forcedt]] and [[#set blockdt|set blockdt]] options | ||
| + | *Integral reaction rates are scored using the collision estimator of neutron flux, which has a few adjustable parameters (see [[#set cfe|set cfe]]). | ||
| + | *For more information on tracking modes, see the detailed description on [[delta- and surface-tracking]]. | ||
| + | |||
| + | === set dynccfile === | ||
| + | |||
| + | '''set dynccfile''' ''OPT'' | ||
| + | Option to store precursors and neutrons between time steps in coupled dynamic simulations into a file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT</tt> | ||
| + | | : option to switch on (1/yes) or off (0/no) the store/write dynamic data into a file. Default is on. | ||
| + | |} | ||
| + | |||
| + | === set dynsrc === | ||
| + | |||
| + | '''set dynsrc''' ''PATH'' [ ''MODE'' ] | ||
| + | |||
| + | Links previously generated steady state source distributions to be used in a transient simulation with delayed neutron emission. | ||
| + | |||
| + | {| | ||
| + | | <tt>''PATH''</tt> | ||
| + | | : The path of the previously generated source file (without the <tt>.main</tt> suffix) | ||
| + | |- | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : Precursor tracking mode (0 = mesh based, 1 = point-wise) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Four source files will be required <tt>[''PATH''].main</tt>, <tt>[''PATH''].prec</tt>, <tt>[''PATH''].live</tt> and <tt>[''PATH''].precpoints</tt> | ||
| + | |||
| + | === set ecut === | ||
| + | |||
| + | '''set ecut''' ''EMIN<sub>n</sub>'' ''EMIN<sub>p<sub>'' | ||
| + | Sets minimum energy cut-off for neutrons and photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''EMIN<sub>n</sub>''</tt> | ||
| + | | : cut-off energy for neutrons (MeV) | ||
| + | |- | ||
| + | | <tt>''EMIN<sub>p</sub>''</tt> | ||
| + | | : cut-off energy for photons (MeV) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The default cut-of energy for photons is 1 keV. Neutron energy cut-off is switched off by default. | ||
| + | *Using energy cut-off for neutrons may lead to non-physical results, since fission and up-scattering may not be accurately modeled. | ||
| + | *Versions 2.1.27 and earlier include only photon energy cut-off, which is now the second input parameter. | ||
| + | |||
| + | === set ecutdens === | ||
| + | |||
| + | '''set ecutdens''' ''DENS<sub>1</sub>'' ''EMIN<sub>p,1</sub>'' [ ''DENS<sub>2</sub>'' ''EMIN<sub>p,2</sub>'' ... ] | ||
| + | Sets density-wise minimum energy cut-off for photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''DENS<sub>i</sub>''</tt> | ||
| + | | : mass density (g/cm<sup>3</sup>) | ||
| + | |- | ||
| + | | <tt>''EMIN<sub>p,i</sub>''</tt> | ||
| + | | : cut-off energy for photons (MeV) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Mass densities and energy cut-offs must be given in ascending order. | ||
| + | |||
| + | === set ecutmat === | ||
| + | |||
| + | '''set ecutmat''' ''MAT<sub>1</sub>'' ''EMIN<sub>p,1</sub>'' [ ''MAT<sub>2</sub>'' ''EMIN<sub>p,2</sub>'' ... ] | ||
| + | Sets material-wise minimum energy cut-off for photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>i</sub>''</tt> | ||
| + | | : material name | ||
| + | |- | ||
| + | | <tt>''EMIN<sub>p,i</sub>''</tt> | ||
| + | | : cut-off energy for photons (MeV) | ||
| + | |} | ||
| + | |||
| + | === set eddi === | ||
| + | |||
| + | '''set eddi''' ''OPT'' | ||
| + | Switches on the calculation of Eddington factors. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch calculation on (1) or off (0) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Requires group constant generation to be set on. | ||
| + | |||
| + | === set edepdel === | ||
| + | |||
| + | '''set edepdel''' ''OPT'' [ ''LOCAL_EGD'' ] | ||
| + | Option to include the energy of delayed components in energy deposition calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : include (1) or exclude (0) the energy of delayed components in energy deposition estimates | ||
| + | |- | ||
| + | | <tt>''LOCAL_EGD''</tt> | ||
| + | | : deposit the energy of the delayed fission gammas to fission sites (1) or with the same distribution as the prompt fission gammas (0) (this option is used only in energy deposition mode 3) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | * Delayed components include delayed neutrons, delayed fission gammas and delayed betas. | ||
| + | * The energy of delayed neutrons can be excluded using this option only in energy deposition mode 1. | ||
| + | * The energy of the delayed components is deposited at the time of fission so the time dependence of the energy deposition is not accounted for properly in transient simulations. | ||
| + | * Default options are to include delayed components (1) and to deposit the energy of the delayed fission gammas with the same distribution as the prompt fission gammas (0). | ||
| + | |||
| + | === set edepkcorr === | ||
| + | |||
| + | '''set edepkcorr''' ''OPT'' | ||
| + | Option to apply correction for energy deposition estimates when simulating non-critical systems with criticality source mode. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : apply (1) or do not apply (0) correction | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | * The methodology is described in related paper. <ref name="edep">Tuominen, R., Valtavirta, V. and Leppänen, J. | ||
| + | ''"New energy deposition treatment in the Serpent 2 Monte Carlo transport code."'' Ann. Nucl. Energy [https://doi.org/10.1016/j.anucene.2019.02.003 '''129''' (2019) 224-232].</ref> | ||
| + | |||
| + | === set edepmode === | ||
| + | |||
| + | '''set edepmode''' ''MODE'' [ ''E_CAPT'' ] | ||
| + | Sets energy deposition mode for energy deposition calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : energy deposition mode (0, 1, 2 or 3) | ||
| + | |- | ||
| + | | <tt>''E_CAPT''</tt> | ||
| + | | : additional energy release in capture reactions given in MeVs per fission (used only in energy deposition mode 1) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | * The energy deposition modes are described in related paper. <ref name="edep" />. | ||
| + | * The choice of energy deposition mode affects also the normalization of the results, if normalization to total power or power density is used. | ||
| + | * Energy deposition modes 1, 2 and 3 require data which is not available in the standard ACE-format cross section files used by Serpent. Separately distributed ACE-files containing additional data are required to use these modes. | ||
| + | * Default option is 0 which corresponds to the methodology used before version 2.1.31. | ||
| + | * KERMA coefficients used in energy deposition modes 2 and 3 are not Doppler-broadened correctly by the built-in preprocessor. See [[Doppler-broadening preprocessor routine|Doppler-broadening preprocessor]]. | ||
| + | |||
| + | === set egrid === | ||
| + | |||
| + | '''set egrid''' ''TOL'' [ ''EMIN EMAX'' ] | ||
| + | |||
| + | Sets the unionized energy grid reconstruction parameters. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TOL''</tt> | ||
| + | | : fractional reconstruction tolerance | ||
| + | |- | ||
| + | | <tt>''EMIN''</tt> | ||
| + | | : minimum energy in the grid (MeV) | ||
| + | |- | ||
| + | | <tt>''EMAX''</tt> | ||
| + | | : maximum energy in the grid (MeV) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default fractional reconstruction tolerance is 0.0 in transport calculation mode and 5E-5 in burnup calculation mode. | ||
| + | *A higher energy grid reconstruction tolerance means lower memory consumption and possibly higher computation speed but also reduced accuracy of the calculation. | ||
| + | *The default minimum energy is 1E-11 MeV (neutrons) and 1E-03 MeV (photons). | ||
| + | *The default maximum energy is 20.0 MeV (neutrons) and 100.0 MeV (photons). | ||
| + | *See also Section 5.3 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set ekn === | ||
| + | |||
| + | '''set ekn''' ''E'' | ||
| + | Sets the Klein-Nishina equation to approximate a Compton scattering event. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''E''</tt> | ||
| + | | : energy cut-off for modelling energy and direction of the scattered photon (MeV) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default value of <tt>''E''</tt> is set to INFTY (1E+37). | ||
| + | *The Klein-Nishina equation is used above <tt>''E''</tt> for calculating both the energy and direction of the scattered photon. Below <tt>''E''</tt>, the Doppler brodening method is used if switched on. Otherwise, the incoherent scattering function approximation is in use. | ||
| + | |||
| + | === set elcond === | ||
| + | |||
| + | '''set elcond''' ''MAT<sub>1</sub>'' ''COND<sub>1</sub>'' [ ''MAT<sub>2</sub>'' ''COND<sub>2</sub>'' ... ] | ||
| + | Sets material-wise conductivity state for electrons in photon transport calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>i</sub>''</tt> | ||
| + | | : material name | ||
| + | |- | ||
| + | | <tt>''COND<sub>i</sub>''</tt> | ||
| + | | : conductivity state (0 = non-conductor, 1 = conductor, 2 = conduction electron dependent). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default option is 2 or conduction electron dependent. | ||
| + | *If the material is set as a conductor and no-conduction electrons are found, the material conductivity state is overwritten to non-conductor. | ||
| + | *Option 2, conduction electron dependent, establishes that a single element material is a conductor if conduction electrons are found, otherwise is a non-conductor. A compound is always a non-conductor. | ||
| + | |||
| + | === set elgas === | ||
| + | |||
| + | '''set elgas''' ''MAT<sub>1</sub>'' ''GAS<sub>1</sub>'' [ ''MAT<sub>2</sub>'' ''GAS<sub>2</sub>'' ... ] | ||
| + | Sets material-wise phase for electrons in photon transport calculations. Input options: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>i</sub>''</tt> | ||
| + | | : material name | ||
| + | |- | ||
| + | | <tt>''GAS<sub>i</sub>''</tt> | ||
| + | | : phase state (0 = condensed or non-gas, 1 = gas). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default option is 0/gas and only affects mixtures. | ||
| + | *The gas phase does not affect the mean excitation energy of a single material: if [[#set elmee|set elmee]] option is set for a material, the material is considered as non-gas. | ||
| + | |||
| + | === set elmee === | ||
| + | |||
| + | '''set elmee''' ''MAT<sub>1</sub>'' ''MEE<sub>1</sub> [ ''MAT<sub>2</sub>'' ''MEE''<sub>2</sub>'' ... ] | ||
| + | Sets material-wise mean excitation energy for electrons in photon transport calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>i</sub>''</tt> | ||
| + | | : material name | ||
| + | |- | ||
| + | | <tt>''MEE<sub>i</sub>''</tt> | ||
| + | | : electrons mean excitation energy (MeV) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The default value is -1.0 interpreted as calculated during runtime for compounds and extracted from data for single element materials. | ||
| + | *The maximum mean excitation energy for electrons is 1 MeV. | ||
| + | |||
| + | === set elspn === | ||
| + | |||
| + | '''set elspn''' ''EGRID_E'' | ||
| + | Sets the stopping power energy grid size for electrons/positrons in photon transport calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''EGRID_E''</tt> | ||
| + | | : energy grid size (default value is 200) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The thick-target bremsstrahlung model ([[Input syntax manual#set ttb|set ttb]] option) assumes that the energy array is uniformly distributed in a log-energy scale. | ||
| + | |||
| + | === set entr === | ||
| + | |||
| + | '''set entr''' ''N<sub>X</sub> N<sub>Y</sub> N<sub>Z</sub>'' [ ''X<sub>MIN</sub> X<sub>MAX</sub> Y<sub>MIN</sub> Y<sub>MAX</sub> Z<sub>MIN</sub> Z<sub>MAX</sub>'' ] | ||
| + | Defines the mesh structure used for calculating fission source entropy. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of mesh cells in x-direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of mesh cells in y-direction | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of mesh cells in z-direction | ||
| + | |- | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : minimum mesh boundary in x-direction | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : maximum mesh boundary in x-direction | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : minimum mesh boundary in y-direction | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : maximum mesh boundary in y-direction | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : minimum mesh boundary in z-direction | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : maximum mesh boundary in z-direction | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Shannon entropy is used to monitor fission source convergence by recording the distribution of source points on mesh. | ||
| + | *The calculation is invoked by setting the generation history record option on ([[#set his|set his]]). | ||
| + | *The default mesh size is 5 × 5 × 5, extending over the entire geometry. | ||
| + | *Monitoring fission source convergence make sense only in criticality source mode. | ||
| + | *For more information, see detailed description on [[fission source convergence]]. | ||
| + | |||
| + | === set fininitfile === | ||
| + | |||
| + | '''set fininitfile''' ''FILEPATH'' | ||
| + | Links a file containing an initial fuel behavior solution used as a starting point for a coupled calculation with the [[FINIX fuel behavior module]]. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILEPATH''</tt> | ||
| + | | : path to the file | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The file should be a [[Multi-physics_interface#Fuel_behavior_interface_.28types_5_and_6.29|type 6 fuel behavior interface]] containing a previous solution from a coupled FINIX calculation. | ||
| + | *The axial and radial nodalization as well as the included fuel rods should be the same in the file as in the calculation. | ||
| + | |||
| + | === set fissh === | ||
| + | |||
| + | '''set fissh''' ''ZAI<sub>1</sub> E<sub>1</sub> ZAI<sub>2</sub> E<sub>2</sub> ...'' | ||
| + | Overrides default fission heating values. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''ZAI<sub>n</sub>''</tt> | ||
| + | | : nuclide identifiers ([[Definitions, units and constants#definitions|ZAI]]) | ||
| + | |- | ||
| + | | <tt>''E<sub>n</sub>''</tt> | ||
| + | | : energy deposited per fission in MeV | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The energy deposited per fission includes additional energy released in capture reactions when fission neutrons are absorbed. | ||
| + | *By default the energy release per U-235 fission is set to 202.27 MeV, and the values for other actinides scaled based the Q-values found in the cross section libraries. | ||
| + | *See also [[Input syntax manual#set U235H|set U235H]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set fissrate === | ||
| + | |||
| + | '''set fissrate''' ''F'' [ ''MAT'' ] | ||
| + | |||
| + | Sets normalization to fission rate. | ||
| + | |||
| + | {| | ||
| + | | <tt>''F''</tt> | ||
| + | | : number of fission reactions per second (1/s) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : dummy parameter | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set powdens|set powdens]], [[#set flux|set flux]], [[#set genrate|set genrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set fissye === | ||
| + | |||
| + | '''set fissye''' ''OPT'' | ||
| + | Option to include energy-dependent fission yields. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : include (1/yes) or exclude (0/no) energy-dependent fission yields. Default is 1. | ||
| + | |} | ||
| + | |||
| + | === set flux=== | ||
| + | |||
| + | '''set flux''' ''F'' [ ''MAT'' ] | ||
| + | Sets normalization to total flux. | ||
| + | |||
| + | {| | ||
| + | | <tt>''F''</tt> | ||
| + | | : flux | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : dummy parameter | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set powdens|set powdens]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set fluxlimtrc=== | ||
| + | |||
| + | '''set fluxlimtrc''' ''OPT'' | ||
| + | Option that enables the calculation of flux limited TRC diffusion coefficients. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch calculation on (1/yes) or off (0/no). Default is off. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *In infinite-spectrum calculations the anisotropic component of the non-TRC nuclides/materials is removed | ||
| + | *S1 and SP1 are non-corrected for non-TRC nuclides/materials and below energy limit for TRC nuclides/materials (see [[#set trc|set trc]] option) | ||
| + | |||
| + | === set fmtx === | ||
| + | |||
| + | '''set fmtx''' '''1''' ''MAT<sub>1</sub>'' ''MAT<sub>2</sub>'' ... | ||
| + | |||
| + | '''set fmtx''' '''2''' ''UNI<sub>1</sub>'' ''UNI<sub>2</sub>'' ... | ||
| + | |||
| + | '''set fmtx''' '''3''' ''LVL'' | ||
| + | |||
| + | '''set fmtx''' '''4''' ''X<sub>MIN</sub>'' ''X<sub>MAX</sub>'' ''N<sub>X</sub>'' ''Y<sub>MIN</sub>'' ''Y<sub>MAX</sub>'' ''N<sub>Y</sub>'' ''Z<sub>MIN</sub>'' ''Z<sub>MAX</sub>'' ''N<sub>Z</sub>'' | ||
| + | |||
| + | Set the calculation of fission matrixes. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>n</sub>''</tt> | ||
| + | | : material list (type 1) | ||
| + | |- | ||
| + | | <tt>''UNI<sub>n</sub>''</tt> | ||
| + | | : universe list (type 2) | ||
| + | |- | ||
| + | | <tt>''LVL''</tt> | ||
| + | | : level number (type 3) | ||
| + | |- | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : minimum x-coordinate mesh boundary (type 4) | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : maximum x-coordinate mesh boundary (type 4) | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of x-mesh cells (type 4) | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : minimum y-coordinate mesh boundary (type 4) | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : maximum y-coordinate mesh boundary (type 4) | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of y-mesh cells (type 4) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : minimum z-coordinate mesh boundary (type 4) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : maximum z-coordinate mesh boundary (type 4) | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of z-mesh cells (type 4) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * There are four options for defining the regions that compose the matrix: 1 = material, 2 = universe, 3 = level, 4 = Cartesian mesh. | ||
| + | * The output is printed in file <tt>[input]_fmtx[bu].m</tt>, where "<tt>bu</tt>" is the burnup step. Output values are given in pairs: matrix element and associated relative error, for total, prompt and delayed (analog) fission matrixes. | ||
| + | |||
| + | === set forcedt === | ||
| + | |||
| + | '''set forcedt''' ''MAT<sub>1</sub> MAT<sub>2</sub> ...'' | ||
| + | Defines the list of materials where delta-tracking is always used. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>n</sub>''</tt> | ||
| + | | : material names | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *This option is used to override selection of tracking mode based on the probability threshold (see [[#set dt|set dt]]) in individual materials. | ||
| + | *Use of delta-tracking can be blocked in individual materials using [[#set blockdt|set blockdt]]. | ||
| + | *For more information on tracking modes, see the detailed description on [[delta- and surface-tracking]]. | ||
| + | *''Note to developers: should have different lists for neutrons and photons?'' | ||
| + | |||
| + | === set fpcut === | ||
| + | |||
| + | '''set fpcut''' ''FPCUT'' | ||
| + | |||
| + | Fission product yield cut-off <tt>''FPCUT''</tt> acts on cumulative fission yields, and the given number is the lower limit for the maximum cumulative yield in each mass chain. So "set fpcut 1E-2" means that every mass chain (nuclides with same mass number) with all cumulative yields below 1% are discarded from the calculation. | ||
| + | |||
| + | The cut-off is set to zero by default. Setting the <tt>''FPCUT''</tt> cut-off to a higher value (~1E-4 or so) is an effective way to reduce the number of nuclides in the calculation, but at some point it will start affecting the results. | ||
| + | |||
| + | === set fsp === | ||
| + | |||
| + | '''set fsp''' ''OPT'' ''NSKIP'' | ||
| + | |||
| + | Sets fission source passing between two transport simulations in burnup or coupled calculation. The fission source at the end of one transport calculation is used as the initial source for the next transport calculation. | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch fission source passing on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''NSKIP''</tt> | ||
| + | | : number of inactive generations on subsequent steps | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Fission source passing is turned off by default. | ||
| + | *Number of inactive generations is taken from [[#set pop|set pop]] card on the first step and from [[#set fsp|set fsp]] on all later steps. | ||
| + | |||
| + | === set fum === | ||
| + | |||
| + | '''set fum''' ''ERG'' [ ''BTCH MODE LIM'' ] | ||
| + | |||
| + | '''set fum''' ''ERG'' [ ''BTCH MODE LIM TGT ITER INIT'' ] | ||
| + | |||
| + | '''set fum''' ''ERG'' [ ''BTCH MODE DC LIM TGT ITER INIT'' ] | ||
| + | |||
| + | Activates fundamental mode calculation for collapsing intermediate multi-group constant data into few-group constants with a critical spectrum. | ||
| + | |||
| + | {| | ||
| + | | <tt>''ERG''</tt> | ||
| + | | : Intermediate multi-group structure for calculation of the leakage-corrected critical spectrum | ||
| + | |- | ||
| + | | <tt>''BTCH''</tt> | ||
| + | | : When set to 2, results are averaged over all criticality cycles | ||
| + | |- | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : Critical spectrum calculation type (default 0, i.e. old B<sub>1</sub> calculation) | ||
| + | |- | ||
| + | | <tt>''DC''</tt> | ||
| + | | : Multi-group diffusion coefficients to be used with FM fundamental mode calculation (default value INF_DIFFCOEF). This value is not read with other modes. | ||
| + | |- | ||
| + | | <tt>''LIM''</tt> | ||
| + | | : Convergence criterion of k<sub>eff</sub> in fundamental mode calculation calculated as the absolute value difference of k<sub>eff</sub> between successive iterations (default value 1E-7) | ||
| + | |- | ||
| + | | <tt>''TGT''</tt> | ||
| + | | : Target value for fundamental mode k<sub>eff</sub> (default value 1.0, not used with the old B<sub>1</sub> calculation mode) | ||
| + | |- | ||
| + | | <tt>''ITER''</tt> | ||
| + | | : Maximum number of fundamental mode calculation iterations (default value 25, not used with the old B<sub>1</sub> calculation mode) | ||
| + | |- | ||
| + | | <tt>''INIT''</tt> | ||
| + | | : First guess for absolute value of critical B<sup>2</sup> (default value 1E-6, not used with the old B<sub>1</sub> calculation mode) | ||
| + | |} | ||
| + | |||
| + | The possible values for mode are: | ||
| + | |||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Mode | ||
| + | ! Description | ||
| + | |- | ||
| + | | <tt>o, O, 0</tt> | ||
| + | | Old B<sub>1</sub> calculation (use the same method as before version 2.1.31) | ||
| + | |- | ||
| + | | <tt>b, B, 1</tt> | ||
| + | | New B<sub>1</sub> calculation | ||
| + | |- | ||
| + | | <tt>p, P, 2</tt> | ||
| + | | P<sub>1</sub> calculation | ||
| + | |- | ||
| + | | <tt>f, F, 3</tt> | ||
| + | | FM calculation | ||
| + | |} | ||
| + | |||
| + | The possible values for FM mode multi-group diffusion coefficients are: | ||
| + | |||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Mode | ||
| + | ! Description | ||
| + | |- | ||
| + | | <tt>1</tt> | ||
| + | | Use out-scattering diffusion coefficients | ||
| + | |- | ||
| + | | <tt>2</tt> | ||
| + | | Use [[#set_trc|transport corrected]] diffusion coefficients | ||
| + | |- | ||
| + | | <tt>3</tt> | ||
| + | | Use [[#set_cmm|cumulative migration method]] diffusion coefficients | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *When invoked, Serpent applies a fundamental mode leakage correction on group constants and produces a [[Output parameters#B1|second set of output parameters]]. | ||
| + | *Before version 2.1.31, the mode parameter was not read, and the old B<sub>1</sub> calculation mode was always used regardless of the parameter given here. | ||
| + | *The group constants are named with B1_ prefix, but they are calculated with fundamental mode calculation type defined with the mode parameter. | ||
| + | *The multi-group structure may be an energy grid defined using the [[#ene (energy grid definition)|ene card]] or a name of a [[pre-defined energy group structures|pre-defined energy group structure]]. | ||
| + | *Setting the fundamental mode calculation overrides the default 70-group structure used for macroscopic data calculation in infinite spectrum. | ||
| + | *Averaging the results over all cycles may improve convergence and speed up the calculation, but all information on statistical errors is lost. | ||
| + | *This option does not affect the flux spectrum used during burnup calculations. | ||
| + | *The calculation modes other than the old B<sub>1</sub> calculation mode were added in version 2.1.31. | ||
| + | *The descriptions of the other methods than the old B<sub>1</sub> method will be presented later in a publication. | ||
| + | *Note that in general the intermediate multi-group structure should have more groups than the few-group structure to get reasonable results for [[#set fum|leakage corrected group constants]] and out-scatter diffusion coefficients. | ||
| + | *Mode 0 (old B<sub>1</sub> calculation) might be very slow with a large intermediate multi-group structure (for example with approximately 2000 groups). Modes 1-3 should run much faster. | ||
| + | |||
| + | === set gbuf === | ||
| + | |||
| + | '''set gbuf''' ''FAC'' [ ''BNK'' ] | ||
| + | Sets the size of photon buffer and event bank. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FAC''</tt> | ||
| + | | : factor (> 1) defining the buffer size | ||
| + | |- | ||
| + | | <tt>''BNK''</tt> | ||
| + | | : event bank size | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Photon buffer refers to pre-allocated memory block used to store photon particle data. This memory is needed for putting secondary photons in que, etc.. | ||
| + | *The buffer factor defines the buffer size relative to simulated batch size. | ||
| + | *The event bank refers to pre-allocated memory block used to store history data on particle events. This bank is used only with certain special options, such as [[importance detectors]] and [[track plotter]]. | ||
| + | *The default values depend on simulation mode, and there is no need to adjust the values unless the calculation terminates with an error. | ||
| + | *''Note to developers: event bank is now the same for both neutrons and photons.'' | ||
| + | |||
| + | === set gct === | ||
| + | |||
| + | '''set gct''' ''OPT'' | ||
| + | Switches on the calculation of group constant statistics test. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch calculation on (1/yes) or off (0/no). Default is off. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *When this option is set, the batch-wise statistical tests are printed in the file <tt>[input]_stat.m</tt>. | ||
| + | *Requires group constant generation to be set on. | ||
| + | *''Note to developers: statistical tests should be documented'' | ||
| + | |||
| + | === set gcu === | ||
| + | |||
| + | '''set gcu''' ''UNI<sub>1</sub>'' [ ''UNI<sub>2</sub>'' ''UNI<sub>3</sub>'' ... ] | ||
| + | Sets the universes for group constant generation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI<sub>n</sub>''</tt> | ||
| + | | : universe where group constants are generated or -1 to switch group constant generation off. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Group constants are by default generated in the root universe (universe 0). | ||
| + | *Group constant generation should be switched off when the results are not needed (this may speed up the calculation). | ||
| + | *See separate description of [[Output parameters#Homogenized group constants|output parameters]]. | ||
| + | *Super-imposed group constant generation universes (i.e. not part of the geometry definition) should not be used so that they cover (partly or totally) the same geometry region as some other group constant generation universe (super-imposed or part of the geometry definition), if group constants are generated only on a single geometry level. | ||
=== set gcut === | === set gcut === | ||
| − | + | '''set gcut''' ''GMAX'' | |
Sets generation cut-off for neutrons. Input values: | Sets generation cut-off for neutrons. Input values: | ||
{| | {| | ||
| − | | <gmax> | + | | <tt>''gmax''</tt> |
| − | | number of simulated generations before cut-off | + | | : number of simulated generations before cut-off |
|} | |} | ||
| − | The generation cut-off can be used in neutron external source simulations, to limit the length of fission chains. | + | <u>Notes:</u> |
| + | *The generation cut-off can be used in neutron external source simulations, to limit the length of fission chains. | ||
| + | *Applicable only to neutron external source simulation (invoked using [[#set nps|set nps]]) | ||
| + | *Generation or time cut-off ([[#set tcut|set tcut]]) is always needed for neutron external source simulations in super-critical systems. | ||
| − | + | === set genrate === | |
| − | * | + | '''set genrate''' ''G'' [ ''MAT'' ] |
| − | * | + | |
| + | Sets normalization to fission neutron generation rate. | ||
| + | |||
| + | {| | ||
| + | | <tt>''G''</tt> | ||
| + | | : number of fission neutrons emitted per second (in neutrons/s) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material in which the fission neutrons are generated | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *If the material name is omitted, the value corresponds to total fission neutron generation rate in the system. | ||
| + | *The neutron generation rate includes only prompt and delayed neutrons emitted in fission. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set powdens|set powdens]], [[#set flux|set flux]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set gpop === | ||
| + | |||
| + | '''set gpop''' ''MAX_HIS'' ''MAX_POP'' ''C'' | ||
| + | Sets the on-the-fly neutron growing population size algorithm. | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAX_HIS''</tt> | ||
| + | | : maximum number of histories | ||
| + | |- | ||
| + | | <tt>''MAX_POP''</tt> | ||
| + | | : maximum population size | ||
| + | |- | ||
| + | | <tt>''C''</tt> | ||
| + | | : parameter to control the population growth | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The on-the-fly neutron growing population size algorithm is used in criticality calculations to accelerate the fission source convergence. | ||
| + | *The growing algorithm methodology is described in related paper. <ref name="gpop">Dufek, J. and Tuttelberg, K. | ||
| + | ''"Monte Carlo criticality calculations accelerated by a growing neutron population."'' Ann. Nucl. Energy [https://doi.org/10.1016/j.anucene.2016.02.015 '''94''' (2016) 16-21].</ref> | ||
| + | |||
| + | === set gsw === | ||
| + | |||
| + | '''set gsw''' ''FILE'' [ ''OPT'' ] | ||
| + | Writes secondary photon source points in coupled neutron-photon transport simulation into a file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : file name where the source points are written | ||
| + | |- | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to include secondary photons in transport calculation (1/yes), (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Applicable only to coupled neutron-photon transport simulation (invoked using [[#set ngamma|set ngamma]]). | ||
| + | *Default photon-production (from neutron reactions) transported simulation option is switched off (0/no). | ||
| + | *Only source points from active cycles are included in criticality source simulations. | ||
| + | |||
| + | === set his === | ||
| + | |||
| + | '''set his''' ''OPT'' | ||
| + | Sets batch history record on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch batch history record on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *When invoked, Serpent collects batch-wise data on k<sub>eff</sub>, fission source entropy etc. and produces a [[Description of output files#History output|separate output file]]. | ||
| + | *Setting the history option also invokes the calculation of fission source (Shannon) entropy. The entropy mesh parameters can be adjusted using [[#set entr|set entr]]. | ||
| + | |||
| + | === set ifp === | ||
| + | |||
| + | '''set ifp''' ''GEN'' | ||
| + | Sets the number of generations to use for calculating the iterated fission probability. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''GEN''</tt> | ||
| + | | : Number of generations. Default value 15. | ||
| + | |} | ||
| + | |||
| + | === set imp === | ||
| + | |||
| + | '''set imp''' ''TYPE'' ''G'' | ||
| + | Defines energy-dependent importances. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : type of dependency (e = energy) | ||
| + | |- | ||
| + | | <tt>''G''</tt> | ||
| + | | : exponential for adjusting importances (default value: -0.5) | ||
| + | |} | ||
| + | |||
| + | '''set imp''' ''TYPE'' ''r<sub>1</sub>'' ''I<sub>1</sub>'' [ ''r<sub>2</sub>'' ''I<sub>2</sub>'' ... ] | ||
| + | Defines geometry-dependent importances for given region(s). Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''TYPE''</tt> | ||
| + | | : type of (geometrical) dependency (c = cell, m = material, u = universe) | ||
| + | |- | ||
| + | | <tt>''r<sub>n</sub>''</tt> | ||
| + | | : region identifier (cell, material or universe name) | ||
| + | |- | ||
| + | | <tt>''I<sub>n</sub>''</tt> | ||
| + | | : importance | ||
| + | |||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Geometry-dependent importances (only) used for variance reduction | ||
| + | *Only cell (geometry-dependent) type importances supported for now | ||
| + | *Geometry-dependent importances requires using surface-tracking (see [[#set dt|set dt]]) | ||
| + | |||
| + | === set impl === | ||
| + | '''set''' '''impl''' ''ICAPT'' [ ''INXN'' ''INUBAR'' ''ILEAK'' ] | ||
| + | |||
| + | Sets implicit reaction modes on or off. | ||
| + | |||
| + | {| | ||
| + | | <tt>''ICAPT''</tt> | ||
| + | | : option to switch implicit capture reactions on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''INXN''</tt> | ||
| + | | : option to switch implicit nxn reactions on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''INUBAR''</tt> | ||
| + | | : number of fission neutrons to emit in each fission (nonzero = implicit treatment, 0 = analog treatment) | ||
| + | |- | ||
| + | | <tt>''ILEAK''</tt> | ||
| + | | : option to switch implicit implicit leakage on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Group constant generation requires implicit nxn reactions to be set on. | ||
| + | *If an implicit nubar is given, the weights of the fission neutrons are scaled to conserve the physical number of fission neutrons. | ||
| + | *Implicit leakage requires group constant generation to be set on. | ||
| + | *See separate description of [[physics options in Serpent]] for differences to other codes. | ||
| + | |||
| + | === set inftrk === | ||
| + | '''set''' '''inftrk''' ''LOOP<sub>n</sub>'' [ ''ERR<sub>n</sub>'' ''LOOP<sub>p</sub>'' ''ERR<sub>p</sub>'' ] | ||
| + | |||
| + | Sets parameters for terminating infinite tracking loops. | ||
| + | |||
| + | {| | ||
| + | | <tt>''LOOP<sub>n</sub>''</tt> | ||
| + | | : number of neutron tracking loops interpreted as a geometry error | ||
| + | |- | ||
| + | | <tt>''ERR<sub>n</sub>''</tt> | ||
| + | | : flag to terminate neutron tracking when an infinite loop occurs (0 = no, 1 = yes) | ||
| + | |- | ||
| + | | <tt>''LOOP<sub>p</sub>''</tt> | ||
| + | | : number of photon tracking loops interpreted as a geometry error | ||
| + | |- | ||
| + | | <tt>''ERR<sub>p</sub>''</tt> | ||
| + | | : flag to terminate photon tracking when an infinite loop occurs (0 = no, 1 = yes) | ||
| + | |- | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Serpent checks for tracking loop length to avoid simulation being stuck in an infinite loop. The simulation is terminated by default if no material is found after the particle has been moved forward 1000000 times. | ||
| + | *Long loops can occur by chance in complicated geometries, and this parameter allows continuing the simulation without terminating with error message. | ||
| + | *Even if the problem can be solved by switching the infinite loop error off, it is advised to check the geometry for possible errors. | ||
| + | |||
| + | === set inventory === | ||
| + | |||
| + | '''set inventory''' ''MAT<sub>1</sub>'' ''MAT<sub>2</sub>'' ... | ||
| + | |||
| + | '''set inventory''' '''top''' ''N'' ''PARA'' | ||
| + | |||
| + | Specifies which nuclides or elements to include in the <tt>[input]_dep.m</tt> output file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>n</sub>''</tt> | ||
| + | | : Nuclide or element to include | ||
| + | |- | ||
| + | | <tt>''N''</tt> | ||
| + | | : Number of nuclides | ||
| + | |- | ||
| + | | <tt>''PARAM''</tt> | ||
| + | | : Parameter name | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *MAT can be: an isotope, element, or a special entry. | ||
| + | *Isotopes are entered using element symbol and mass number (e.g U-235, Am-242m, etc.) or ZAI (922350, 952421, etc.). In the ZAI format the last digit refers to the isomeric state (0 = ground state, 1 = isomeric state). | ||
| + | *Elements are entered using symbol or numerical (U, 92, etc.). The output includes the sum over all isotopes. | ||
| + | *Special entries include: | ||
| + | **"all" -- All nuclides. | ||
| + | **"accident" -- Nuclides with significant health impact & high migration probability in accident conditions. Source [http://www.oecd-nea.org/rp/chernobyl/c02.html Chernobyl: Assessment of Radiological and Health Impact] | ||
| + | **"actinides" -- Actinides (Z>88) for which cross section data are found in JEFF-3.1.1 | ||
| + | **"burnupcredit" -- Nuclides commonly considered in burnup credit criticality analyses for PWR fuels [http://www.oecd-nea.org/science/wpncs/ADSNF/SOAR_final.pdf] | ||
| + | **"burnupindicators" -- Burnup indicators (commonly measured from spent fuel) [http://www.oecd-nea.org/science/wpncs/ADSNF/SOAR_final.pdf] | ||
| + | ** "cosi6" -- Inventory list used by the COSI6 code (excluding lumped fission products) | ||
| + | ** "lanthanides" -- Lanthanides (56<Z<72) for which cross section data are found in JEFF-3.1.1 | ||
| + | ** "longterm" -- Relevant radionuclides in long-term waste analyses <ref>"''The identification of radionuclides relevant to long-term waste management in th United Kingdom''", Nirex Report no. N/105 (2004)</ref> | ||
| + | ** "minoractinides" -- Minor actinides (actinides - thorium - uranium - plutonium) for which cross section data are found in JEFF-3.1.1 | ||
| + | ** "fp" -- Fission products | ||
| + | ** "dp" -- Actinide decay products | ||
| + | ** "ng" -- Noble gases | ||
| + | *The second syntax allows listing the most significant contributors to a given result. The parameters are: | ||
| + | **"mass" -- contribution to mass fraction | ||
| + | **"activity" -- contribution to activity | ||
| + | **"sf" -- contribution to spontaneous fission rate | ||
| + | **"gsrc" -- contribution to gamma emission rate | ||
| + | **"dh" -- contribution to decay heat | ||
| + | **"ingtox" -- contribution to ingestion toxicity | ||
| + | **"inhtox" -- contribution to inhallation toxicity | ||
| + | *for example "top 10 dh" gives the top 10 contributors to decay heat. | ||
| + | *Since most significant contributors may change over time, the output may contain more nuclides than is requested in the input card. | ||
| + | *The calculation of top contributors does not work with the "-rdep" command line option. | ||
| + | |||
| + | === set isobra === | ||
| + | |||
| + | '''set isobra''' ''ZAI<sub>1</sub>'' ''MT<sub>1</sub>'' ''FG<sub>1</sub>'' | ||
| + | ''ZAI<sub>2</sub>'' ''MT<sub>2</sub>'' ''FG<sub>2</sub>'' | ||
| + | ... | ||
| + | Defines constant branching ratios to isomeric states. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''ZAI<sub>n</sub>''</tt> | ||
| + | | : nuclide identifiers ([[Definitions, units and constants#definitions|ZAI]]) | ||
| + | |- | ||
| + | | <tt>''MT<sub>n</sub>''</tt> | ||
| + | | : [[ENDF reaction MT's and macroscopic reaction numbers#ENDF Reaction MT's|ENDF reaction MT]] | ||
| + | |- | ||
| + | | <tt>''FG<sub>n</sub>''</tt> | ||
| + | | : fraction of reactions leading to the ground state of the product nuclide | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Serpent uses [[Default isomeric branching ratios|constant branching ratios]] by default. This option overrides the default values. | ||
| + | *Energy-dependent data read read from ENDF format files defined by the [[#set bralib|set bralib]] overrides the constant ratios. | ||
| + | |||
| + | === set iter alb === | ||
| + | |||
| + | '''set''' '''iter''' '''alb''' [ ''CYCLES'' ''KEFF'' ''F<sub>X</sub>'' ''F<sub>Y</sub>'' ''F<sub>Z</sub>'' ] | ||
| + | |||
| + | Option that iterates the albedo boundary conditions given a target k-eff for the evaluation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''CYCLES''</tt> | ||
| + | | : number of additional inactive cycles to run for the convergence of the iteration (default value: 50) | ||
| + | |- | ||
| + | | <tt>''KEFF''</tt> | ||
| + | | : target k<sub>eff</sub> for the iteration (default value: 1.0) | ||
| + | |- | ||
| + | | <tt>''F<sub>X</sub>''</tt> | ||
| + | | : albedo factor in x-direction to normalize the albedo leakage rate (default value: 1.0) | ||
| + | |- | ||
| + | | <tt>''F<sub>Y</sub>''</tt> | ||
| + | | : albedo factor in y-direction to normalize the albedo leakage rate (default value: 1.0) | ||
| + | |- | ||
| + | | <tt>''F<sub>Z</sub>''</tt> | ||
| + | | : albedo factor in z-direction to normalize the albedo leakage rate (default value: 1.0) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The albedo boundary conditions result from the product of the iterated albedo leakage rate by the albedo factor in each direction. | ||
| + | |||
| + | === set iter nuc === | ||
| + | |||
| + | {{:Critical density iteration}} | ||
| + | *See [[Critical density iteration]]. | ||
| + | |||
| + | === set keff === | ||
| + | |||
| + | '''set keff''' ''K<sub>EFF</sub>'' | ||
| + | Option to scale fission neutron production in external source simulations. | ||
| + | |||
| + | {| | ||
| + | | <tt>''K<sub>EFF</sub>''</tt> | ||
| + | | : Fission neutron production scaling factor. Default value is 1. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The k-effective to use for scaling the fission neutron production. Inverse of <tt>''K<sub>EFF</sub>''</tt> will be used as a multiplicative constant for the nubar, i.e. a value of 2.0 will cut the fission neutron production in half. | ||
| + | *Affects both prompt and delayed neutron production from fissions. | ||
| + | *In versions '''prior to 2.1.31''': does not affect delayed neutron precursor production, which will cause unexpected behaviour in [[Transient simulations]] that track delayed neutron precursor concentrations. | ||
| + | |||
| + | === set lossrate === | ||
| + | |||
| + | '''set lossrate''' ''L'' [ ''MAT'' ] | ||
| + | |||
| + | Sets normalization to total loss rate. | ||
| + | |||
| + | {| | ||
| + | | <tt>''L''</tt> | ||
| + | | : number of lost neutrons per second (neutrons/s) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : dummy parameter | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *Loss rate includes absorption rate and leakage. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set powdens|set powdens]], [[#set powdens|set flux]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set lost === | ||
| + | |||
| + | '''set lost''' ''LIM'' | ||
| + | Option to treat undefined geometry regions as void. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIM''</tt> | ||
| + | | : maximum number of collisions allowed in undefined regions or -1 if no limit is set. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option allows the calculation to proceed even if the geometry routine encounters undefined cells. | ||
| + | *The option is intended, for example, for complex geometries in which the boundaries of adjacent cells may not fully coincide. | ||
| + | *When the option is set, the number of lost particles is printed in variable LOST_PARTICLES in the <tt>[input]_res.m</tt> output file. | ||
| + | *The option should not be used to get away with errors in incomplete or poorly defined geometries. | ||
| + | |||
| + | === set maxsplit === | ||
| + | |||
| + | '''set maxsplit''' ''MAX'' ''MIN'' | ||
| + | Sets limiting values for splitting and Russian roulette. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAX''</tt> | ||
| + | | : maximum number of splits | ||
| + | |- | ||
| + | | <tt>''MIN''</tt> | ||
| + | | : minimum survival probability in Russian roulette | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Used with weight-windows (see [[Input_syntax_manual#wwin (weight window mesh definition)|weight window mesh definition]]). | ||
| + | |||
| + | === set mbtch === | ||
| + | |||
| + | '''set mbtch''' ''N'' | ||
| + | Adjusts the batch used with MPI data transfer. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''N''</tt> | ||
| + | | : batch size in double floats (8 bytes) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The batch size determines the division of data blocks in MPI data transfer. The value may have some effect on parallel performance. | ||
| + | *The batch size is set to 10000 by default. | ||
| + | |||
| + | === set mcvol === | ||
| + | |||
| + | '''set mcvol''' ''NP'' | ||
| + | Runs the Monte Carlo volume-checker routine to set material volumes before running the transport simulation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NP''</tt> | ||
| + | | : number of points sampled in the geometry | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The [[Installing and running Serpent#Monte Carlo volume calculation routine|Monte Carlo based volume calculation routine]] works by sampling random points in the geometry, and counting the number of hits in every material. | ||
| + | *When invoked, all material volumes are overridden by the results given by the checker routine. | ||
| + | *The volume checker can also be used to produce a separate input file for the volume entries (see detailed description on [[defining material volumes]]). | ||
| + | |||
| + | === set mdep === | ||
| + | |||
| + | '''set mdep''' ''UNI VOL N MAT<sub>1</sub> MAT<sub>2</sub> ... MAT<sub>N</sub> '' | ||
| + | ''ZAI<sub>1</sub>'' ''MT<sub>1</sub> | ||
| + | ''ZAI<sub>2</sub>'' ''MT<sub>2</sub> | ||
| + | ... | ||
| + | Sets parameters for calculating homogenized microscopic cross sections. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe where universe where homogenized microscopic cross sections are generated | ||
| + | |- | ||
| + | | <tt>''VOL''</tt> | ||
| + | | : volume of the universe | ||
| + | |- | ||
| + | | <tt>''N''</tt> | ||
| + | | : number of materials included in the calculation | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub> MAT<sub>2</sub> ... MAT<sub>N</sub>''</tt> | ||
| + | | : material names | ||
| + | |- | ||
| + | | <tt>''ZAI<sub>i</sub>''</tt> | ||
| + | | : nuclide identifier ([[Definitions, units and constants#definitions|ZAI]]) | ||
| + | |- | ||
| + | | <tt>''MT<sub>i</sub>''</tt> | ||
| + | | : ENDF reaction [[ENDF reaction MT's and macroscopic reaction numbers|MT]] | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *When this option is set, Serpent calculates homogenized few-group microscopic cross sections for the listed nuclides and reactions. | ||
| + | *The cross sections are always calculated in the actual spectrum of the problem, never with the [[#set fum|critical spectrum]]. | ||
| + | *The results are printed in a [[Description of output files#Micro depletion output|separate output file]]. | ||
| + | *The results can be included in the [[Description_of_output_files#Group_constant_output|group constant output]] by adding <tt>MDEP_XS</tt> in the [[#set_coefpara|set coefpara]] list. | ||
| + | *If the number of materials is zero, the calculation is carried over all burnable materials. | ||
| + | *If the list of nuclides and reactions is substituted by "all", Serpent will generate the micro-depletion output including all the nuclides and all reactions involved in the calculation at beginning of the simulation aiming to the extract the constant data (stopping the calculation right after). | ||
| + | *The listed materials must be enclosed inside the homogenized universe. | ||
| + | *The calculation requires the [[Defining material volumes|material volumes to be correctly set]]. | ||
| + | *Transmutation reactions to ground and isomeric states can be calculated by adding 'g' and 'm' after the reaction MT (e.g. 102m is the capture cross section corresponding to daughter nuclide being in isomeric state). Reaction rates are calculated to all states by default. | ||
| + | *Fission reactions corresponding to a specific yield in ENDF can be calculated by adding 1, 2, 3, 4, ... after the reaction MT (e.g. 181 is total fission cross section corresponding to first fission product yield) depending on the fission yield data of the nuclide in the data library. This parameter was different before 2.1.32. | ||
| + | *Sum of all capture reactions can be obtained using MT 101 | ||
| + | *Some actinides are missing the total fission channel, and setting the MT to 18 produces sum over MT's 19, 20, 21 and 38 (from version 2.1.29 on). | ||
| + | *The nuclide identifiers are entered as ZAI, not ZA. For example, the ZAI for U-235 is 922350 and the ZAI for Am-242m is 952421. | ||
| + | *Parameter ''VOL'' was ''VR'' in versions before 2.1.32 (volume ratio of materials included in micro depletion to total homogenized region). | ||
| + | *Multiple set mdep cards can be given for a single homogenized universe. | ||
| + | *The microscopic cross section calculation methodology is described in a related article.<ref>Rintala, A., Valtavirta, V. and Leppänen, J. ''"Microscopic cross section calculation methodology in the Serpent 2 Monte Carlo code."'' Ann. Nucl. Energy [https://doi.org/10.1016/j.anucene.2021.108603 '''164''' (2021) 108603]</ref> | ||
| + | |||
| + | === set memfrac === | ||
| + | |||
| + | '''set memfrac''' ''FRAC'' | ||
| + | |||
| + | Defines the fraction of total system memory Serpent can allocate to its use. If the fraction is exceeded, the simulation will abort. This is mainly to avert the use of swap-memory, which can make the system unresponsive. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FRAC''</tt> | ||
| + | | : the fraction of system memory that Serpent is allowed to use (between 0 and 1) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * The default fraction is 0.8. | ||
| + | * The fraction can also be set via a SERPENT_MEM_FRAC environmental variable. | ||
| + | * Serpent tries to read the system total memory from the first line of the file /proc/meminfo | ||
| + | |||
| + | === set mfpcut === | ||
| + | |||
| + | '''set mfpcut''' ''MFPMIN<sub>p</sub>'' | ||
| + | Sets minimum mean-free-path cut-off for photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MFPMIN<sub>p</sub>''</tt> | ||
| + | | : cut-off mean-free-path for photons (cm) | ||
| + | |} | ||
| + | |||
| + | === set mfpcutdens === | ||
| + | |||
| + | '''set mfpcutdens''' ''DENS<sub>1</sub>'' ''MFPMIN<sub>p,1</sub>'' [ ''DENS<sub>2</sub>'' ''MFPMIN<sub>p,2</sub>'' ... ] | ||
| + | Sets density-wise minimum mean-free-path cut-off for photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''DENS<sub>i</sub>''</tt> | ||
| + | | : mass density (g/cm<sup>3</sup>) | ||
| + | |- | ||
| + | | <tt>''MFPMIN<sub>p,i</sub>''</tt> | ||
| + | | : cut-off mean-free-path for photons (cm) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Mass densities and mean-free-paths cut-offs must be given in ascending order. | ||
| + | |||
| + | === set mfpcutmat === | ||
| + | |||
| + | '''set mfpcutmat''' ''MAT<sub>1</sub>'' ''MFPMIN<sub>p,1</sub>'' [ ''MAT<sub>2</sub>'' ''MFPMIN<sub>p,2</sub>'' ... ] | ||
| + | Sets material-wise minimum mean-free-path cut-off for photons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>i</sub>''</tt> | ||
| + | | : material name | ||
| + | |- | ||
| + | | <tt>''MFPMIN<sub>p,i</sub>''</tt> | ||
| + | | : cut-off mean-free-path for photons (cm) | ||
| + | |} | ||
| + | |||
| + | === set micro === | ||
| + | |||
| + | '''set micro''' ''ERG'' [ ''BTCH'' ] | ||
| + | Defines the intermediate multi-group structure used for group constant generation. | ||
| + | |||
| + | {| | ||
| + | | <tt>''ERG''</tt> | ||
| + | | : Intermediate multi-group structure used for group constant generation | ||
| + | |- | ||
| + | | <tt>''BTCH''</tt> | ||
| + | | : When set to 2, results are averaged over all criticality cycles | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Serpent uses an intermediate multi-group structure to calculate [[Output parameters#Homogenized group constants|homogenized few-group constants]]. This input parameter is used to override the default 70-group structure used in the calculation. | ||
| + | *The multi-group structure may be an energy grid defined using the [[#ene (energy grid definition)|ene card]] or a name of a [[pre-defined energy group structures|pre-defined energy group structure]]. | ||
| + | *Averaging the results over all cycles may improve convergence and speed up the calculation, but all information on statistical errors is lost. | ||
| + | *Note that in general the intermediate multi-group structure should have more groups than the few-group structure to get reasonable results for [[#set fum|leakage corrected group constants]] and out-scatter diffusion coefficients. | ||
| + | |||
| + | === set minxs === | ||
| + | |||
| + | See [[#set cfe|set cfe]]. | ||
| + | |||
| + | === set multilevelgcu === | ||
| + | |||
| + | '''set multilevelgcu''' ''OPT'' | ||
| + | Option that enables the generation of group constants in multiple overlapping universes. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch calculation on (1/yes) or off (0/no). Default is off. | ||
| + | |} | ||
| + | |||
| + | === set mvol === | ||
| + | |||
| + | '''set mvol''' ''MAT<sub>1</sub> ZONE<sub>1,1</sub> VOL<sub>1,1</sub>'' | ||
| + | ''MAT<sub>1</sub> ZONE<sub>1,2</sub> VOL<sub>1,2</sub>'' | ||
| + | ... | ||
| + | ''MAT<sub>2</sub> ZONE<sub>2,1</sub> VOL<sub>2,1</sub>'' | ||
| + | ... | ||
| + | Sets the volumes of material regions. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT<sub>m</sub>''</tt> | ||
| + | | : name of material ''m'' | ||
| + | |- | ||
| + | | <tt>''ZONE<sub>m,n</sub>''</tt> | ||
| + | | : index of zone ''n'' in material ''m'' | ||
| + | |- | ||
| + | | <tt>''VOL<sub>m,n</sub>''</tt> | ||
| + | | : volume of zone ''n'' in material ''m'' | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *This option is used to define material volumes manually. The input card is also produced when the [[Installing and running Serpent#Monte Carlo volume calculation routine|Monte Carlo based volume checker routine]] is invoked. | ||
| + | *The zone index is related to [[automated depletion zone division]], invoked by the [[#div (divisor definition)|div]] card. If no division is used, the index must be set to zero for non-burnable materials. For burnable materials the indexing starts from 1. If no division is used, the corresponding index can be found using the [[Installing and running Serpent#Monte Carlo volume calculation routine|Monte Carlo based volume checker routine]]. | ||
| + | *Another option to define material volumes is to use the vol entry in the [[#mat (material definition)|material card]]. | ||
| + | *For more infomation, see detailed description on [[Defining material volumes|the definition of material volumes]]. | ||
| + | |||
| + | === set nbuf === | ||
| + | |||
| + | '''set nbuf''' ''FAC'' [ ''BNK'' ] | ||
| + | Sets the size of neutron buffer and event bank. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FAC''</tt> | ||
| + | | : factor (> 1) defining the buffer size | ||
| + | |- | ||
| + | | <tt>''BNK''</tt> | ||
| + | | : event bank size | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Neutron buffer refers to pre-allocated memory block used to store neutron particle data. This memory is needed for banking fission neutrons for the next generation and putting secondary neutrons in que. | ||
| + | *The buffer factor defines the buffer size relative to simulated batch size. | ||
| + | *The event bank refers to pre-allocated memory block used to store history data on particle events. This bank is used only with certain special options, such as [[importance detectors]] and [[track plotter]]. | ||
| + | *The default values depend on simulation mode, and there is no need to adjust the values unless the calculation terminates with an error. | ||
| + | *With a large [[#set pop|number of neutrons per generation]], lowering the factor might be necessary due to excessive memory usage. The default values of the factor allows for large fluctuations of the neutron population due to poor statistics, which is seldom an issue with larger neutron populations. | ||
| + | *''Note to developers: event bank is now the same for both neutrons and photons.'' | ||
| + | |||
| + | === set nfg === | ||
| + | |||
| + | '''set nfg''' ''ERG'' | ||
| + | |||
| + | Defines the few-group structure used for group constant generation. | ||
| + | |||
| + | {| | ||
| + | | <tt>''ERG''</tt> | ||
| + | | : Name of the few-group structure used for group constant generation | ||
| + | |} | ||
| + | |||
| + | '''set nfg''' ''NE'' | ||
| + | |||
| + | Defines the few-group structure used for group constant generation. This syntax should not be used anymore. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NE''</tt> | ||
| + | | : Number of energy groups (1/2/4) corresponding to Serpent 1 default 1-, 2- or 4-group structure. | ||
| + | |} | ||
| + | |||
| + | '''set nfg''' ''NE E<sub>NE-1</sub> E<sub>NE-2</sub> ... E<sub>1</sub>'' | ||
| + | |||
| + | Defines the few-group structure used for group constant generation. This syntax should not be used anymore. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NE''</tt> | ||
| + | | : Number of energy groups. Must be at least 2 in this format. | ||
| + | |- | ||
| + | | <tt>''E<sub>N</sub>''</tt> | ||
| + | | : Energy group boundary value between groups ''N'' and ''N+1''. Values have to be given in ascending order. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The few-group structure may be an energy grid defined using the [[#ene (energy grid definition)|ene card]] or a name of a [[pre-defined energy group structures|pre-defined energy group structure]]. | ||
| + | *The default is a two-group structure with boundary between fast and thermal group set to 0.625 eV. | ||
| + | *Serpent uses an intermediate multi-group structure in the calculation. The default structure consists of 70 groups, and can be changed using the [[#set micro|set micro]] or [[#set fum|set fum]] options. | ||
| + | *Note that in general the intermediate multi-group structure should have more groups than the few-group structure to get reasonable results for [[#set fum|leakage corrected group constants]] and out-scatter diffusion coefficients. | ||
| + | *The few-group structure must be a sub-set of the intermediate multi-group structure. | ||
| + | *See also: [[Output parameters#Homogenized group constants|group constant output]]. | ||
| + | *The few-group structure will also be used for example for [[#set adf|assembly discontinuity factors]] and [[#set alb|albedos]]. | ||
| + | |||
| + | === set nfylib === | ||
| + | |||
| + | '''set nfylib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets the neutron-induced fission yield library file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : library file paths | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Fission yield libraries are standard ENDF format files containing neutron-induced fission yield data. | ||
| + | *If the file path contains special characters it is advised to enclose it within quotes. | ||
| + | *A default directory path can be set by defining environment variable <tt>SERPENT_DATA</tt>. The code looks for fission yield data files in this path if not found at the absolute. | ||
| + | *From version 2.2.0 and on, a default neutron-induced fission yield library directory file can be set by defining environment variable <tt>SERPENT_NFYLIB</tt>. This file will be used if no other path is given with '''set nfylib'''. | ||
| + | |||
| + | === set ngamma === | ||
| + | |||
| + | '''set ngamma''' ''MODE'' [ ''WMIN'' ''NMAX'' ] | ||
| + | Sets the coupled neutron-photon transport simulation on. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : simulation mode (0 = off, 1 = analog, 2 = implicit) | ||
| + | |- | ||
| + | | <tt>''WMIN''</tt> | ||
| + | | : weight limit for implicit mode (default value: 0.1) | ||
| + | |- | ||
| + | | <tt>''NMAX''</tt> | ||
| + | | : maximum number of emitted photons in implicit mode (default value: 10) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *This input card invokes the production of prompt gammas in neutron reactions. The coupled simulation mode requires that both neutron and photon data libraries are defined. | ||
| + | *In analog mode, the average number of secondary photons produced per collision is defined by the ratio of photon production cross section to material total. Each emitted photon assumes the weight of the incident neutron. | ||
| + | *The implicit mode can be used to produce more photons by allowing variation in their statistical weight. The weight limit defines the minimum allowed weight of emitted photons. This method is close to what is used in MCNP. | ||
| + | *Both calculation modes produce photons in all collisions without any correlation to the sampled reaction mode. | ||
| + | *The <tt>''MODE''</tt> option was incorrectly described until Dec. 12, 2017. | ||
| + | |||
| + | === set nphys === | ||
| + | |||
| + | '''set nphys''' ''FISS'' [ ''CAPT'' ''SCATT'' ] | ||
| + | Option to set reaction modes for neutrons on and off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FISS''</tt> | ||
| + | | : option to handle fission (0 = not handled, 1 = handled) | ||
| + | |- | ||
| + | | <tt>''CAPT''</tt> | ||
| + | | : option to handle capture (0 = not handled, 1 = handled) | ||
| + | |- | ||
| + | | <tt>''SCATT''</tt> | ||
| + | | : option to handle scattering (0 = not handled, 1 = handled) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *All reaction modes are handled by default. | ||
| + | *If fission is switched off, it is handled as capture. | ||
=== set nps === | === set nps === | ||
| − | + | '''set nps''' ''PP'' [ ''BTCH TBI'' ] | |
Sets parameters for simulated particle population in external source mode. Input values: | Sets parameters for simulated particle population in external source mode. Input values: | ||
{| | {| | ||
| − | | < | + | | <tt>''PP''</tt> |
| − | | number of particles | + | | : total number of particles |
|- | |- | ||
| − | | < | + | | <tt>''BTCH''</tt> |
| − | | number of batches | + | | : number of batches |
| + | |- | ||
| + | | <tt>''TBI''</tt> | ||
| + | | : time binning for dynamic mode | ||
|} | |} | ||
| − | + | <u>Notes:</u> | |
| − | + | ||
| − | < | + | |
| − | *Using the nps card sets the mode to external source simulation. Criticality source simulation for neutrons is invoked using [[#set pop|set pop]]. | + | *The total number of particles is divided by the given number of batches to give the number of particles per batch. |
| + | *Default number of batches is 200. | ||
| + | *Using the nps card sets the mode to external source simulation. Criticality source simulation for neutrons is invoked using [[#set pop|set pop]]. (The two cards are mutually exclusive). | ||
| + | *If time binning is provided, the simulation is run in the [[Dynamic external source simulation mode|dynamic mode]] with sequential population control. The bin structure is defined using the [[#tme (time binning definition)|tme card]]. | ||
*Running an external source simulation requires a source, defined by the [[#src|src card]]. Source definition also sets the transported particle type. | *Running an external source simulation requires a source, defined by the [[#src|src card]]. Source definition also sets the transported particle type. | ||
| − | *Neutron external source simulations are limited to sub-critical systems, unless time cut-off ([[#set tcut|set tcut]]) or generation cut-off ([[#set gcut|set gcut]]) is invoked. | + | *Neutron external source simulations are limited to sub-critical systems, unless dynamic mode, time cut-off ([[#set tcut|set tcut]]) or generation cut-off ([[#set gcut|set gcut]]) is invoked. |
*Neutron external source simulations in multiplying systems may require adjusting the neutron buffer ([[#set nbuf|set nbuf]]). | *Neutron external source simulations in multiplying systems may require adjusting the neutron buffer ([[#set nbuf|set nbuf]]). | ||
| − | *Delayed neutron emission is switched off by default in neutron external source simulation | + | *Delayed neutron emission is switched off by default in neutron external source simulation. Delayed neutrons can be included with [[#set delnu|set delnu]]. |
| + | *In transient simulations, where an initial transient source is linked using the [[#set dynsrc|set dynsrc]] option, <tt>''PP''</tt> particles are sampled for each time interval. | ||
| + | |||
| + | === set opti === | ||
| + | |||
| + | '''set opti''' ''MODE'' | ||
| + | |||
| + | Sets the optimization mode which affects the performance and memory usage. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : optimization mode | ||
| + | |} | ||
| + | |||
| + | The possible settings for mode are: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! <tt>''MODE''</tt> | ||
| + | ! Description | ||
| + | |- | ||
| + | | <tt>1</tt> | ||
| + | | Minimum optimization and small memory usage. Suitable for very large burnup calculation problems involving tens or hundreds of thousands of depletion zones. | ||
| + | |- | ||
| + | | <tt>2</tt> | ||
| + | | Good performance in burnup calculations involving several thousand depletion zones. Suitable for research reactor applications, but not the best choice for group constant generation. | ||
| + | |- | ||
| + | | <tt>3</tt> | ||
| + | | Similar to mode 4, but lower memory demand. CPU time required by burnup and processing routines increases steeply along with the number of depletion zones, which makes the mode better suited for small burnup calculation problems. | ||
| + | |- | ||
| + | | <tt>4</tt> | ||
| + | | Maximum performance at the cost of memory usage. Suitable for group constant generation and 2D assembly burnup calculations with a limited number of depletion zones. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Default value for the optimization mode is 4. | ||
| + | *The mode 4 is essentially the same as the methodology in Serpent 1. | ||
| + | *The methodology is described in a paper presented in PHYSOR 2012<ref name="opti">Leppänen, J. and Isotalo, A. ''"Burnup calculation methodology in the Serpent 2 Monte Carlo code."'' In proc. PHYSOR 2012, Knoxville, TN, Apr. 15-20, 2012.</ref>. | ||
| + | |||
| + | === set outp === | ||
| + | |||
| + | '''set outp''' ''INT'' | ||
| + | Sets the interval (in cycles) for writing simulation output to files. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''INT''</tt> | ||
| + | | : number of cycles after which the output-files are updated | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Default value is 50. | ||
| + | *In coupled transient simulations the interval refers to time steps rather than batches. By default, the output is written after every 50th time step. | ||
| + | *Affects files such as <tt>[input]_res.m</tt> and <tt>[input]_det.m</tt> as well as mesh plots. | ||
| + | |||
| + | === set pbuf === | ||
| + | |||
| + | '''set pbuf''' ''FAC'' | ||
| + | Sets the size of the precursors buffer. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FAC''</tt> | ||
| + | | : factor (>1) defining the buffer size | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Precursor buffer refers to pre-allocated memory block used to store precursors data. This memory is needed for banking/retrieving precursors for the next generation/time-interval. | ||
| + | *The buffer factor defines the buffer size relative to simulated batch/cycle size. | ||
| + | |||
| + | === set pcc === | ||
| + | '''set pcc''' ''MODE'' [ ''SSP SSC'' ] | ||
| + | |||
| + | '''set pcc''' '''5''' ''PRED'' ''CORR'' [ ''SSP SSC'' ] | ||
| + | |||
| + | Sets the time integration method in burnup calculation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : time integration method | ||
| + | |- | ||
| + | | <tt>''PRED''</tt> | ||
| + | | : predictor step integration scheme (0 = constant extrapolation, 1 = linear extrapolation) | ||
| + | |- | ||
| + | | <tt>''CORR''</tt> | ||
| + | | : corrector step integration scheme (1 = linear interpolation, 2 = quadratic interpolation) | ||
| + | |- | ||
| + | | <tt>''SSP''</tt> | ||
| + | | : number of substeps for predictor steps | ||
| + | |- | ||
| + | | <tt>''SSC''</tt> | ||
| + | | : number of substeps for corrector steps | ||
| + | |} | ||
| + | |||
| + | The possible settings for mode are: | ||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Mode | ||
| + | ! Predictor method | ||
| + | ! Corrector method | ||
| + | |- | ||
| + | | <tt>0, CE</tt> | ||
| + | | Constant extrapolation | ||
| + | | - | ||
| + | |- | ||
| + | | <tt>1, CELI</tt> | ||
| + | | Constant extrapolation | ||
| + | | Linear interpolation | ||
| + | |- | ||
| + | | <tt>2, LE</tt> | ||
| + | | Linear extrapolation | ||
| + | | - | ||
| + | |- | ||
| + | | <tt>3, LELI</tt> | ||
| + | | Linear extrapolation | ||
| + | | Linear interpolation | ||
| + | |- | ||
| + | | <tt>4, LEQI</tt> | ||
| + | | Linear extrapolation | ||
| + | | Quadratic interpolation | ||
| + | |- | ||
| + | | <tt>5</tt> | ||
| + | | Constant or linear extrapolation | ||
| + | | Linear or quadratic interpolation | ||
| + | |- | ||
| + | | <tt>6, CECE</tt> | ||
| + | | Constant extrapolation | ||
| + | | Constant backwards extrapolation | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *<tt>''MODE''</tt> 0 is also known as "Euler's method". | ||
| + | *<tt>''MODE''</tt> 1 is also known as the "old predictor-corrector method". | ||
| + | *<tt>''MODE''</tt> 0 and 1 without substeps corresponds to the methods used in Serpent 1. | ||
| + | *The default numbers of predictor and corrector substeps (<tt>''SSP''</tt> and <tt>''SSC''</tt>, respectively) are 1 for applicable methods. | ||
| + | *The default time integration scheme corresponds to <tt>''MODE''</tt> 1 for applicable methods. | ||
| + | *Number of substeps could not be given for constant predictor or corrector before 2.1.32. | ||
| + | *Decay calculations were always calculated with single substep disregarding this input before 2.1.32. | ||
| + | *The first burnup step extrapolation is always constant and with only single substeps before 2.1.32. | ||
| + | *The custom mode, <tt>''MODE''</tt> '''5''', accepts constant or linear extrapolation for the predictor step and linear or quadratic interpolation for the correct step. | ||
| + | |||
| + | === set pdatadir === | ||
| + | |||
| + | '''set pdatadir''' ''DIR'' | ||
| + | Sets the file path for auxiliary photon data. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''DIR''</tt> | ||
| + | | : file path for directory where the data is located | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Serpent uses auxiliary data files for the modelling of photon interaction physics. | ||
| + | *For more information, see [http://serpent.vtt.fi/mediawiki/index.php/Installing_and_running_Serpent#Setting_up_the_data_libraries instructions on setting up the data libraries]. | ||
| + | |||
| + | === set poi === | ||
| + | |||
| + | '''set poi''' ''OPT'' ''VOL'' [ ''XE135M'' ] | ||
| + | Switches the calculation of poison cross sections on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch the calculation of poison cross sections on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''VOL''</tt> | ||
| + | | : volume of the homogenized zone | ||
| + | |- | ||
| + | | <tt>''XE135M''</tt> | ||
| + | | : option to treat <sup>135m</sup>Xe separate from ground state <sup>135</sup>Xe (0 = lumped with <sup>135</sup>Xe, 1 = separate treatment) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Poison cross sections include the fission yields and microscopic and macroscopic absorption cross sections of fission product poisons <sup>135</sup>Xe and <sup>149</sup>Sm, as well as the fission yields and microscopic absorption cross sections of their precursors. The data is part of the [[Output parameters#Homogenized group constants|homogenized group constant output]]. | ||
| + | *The calculation requires setting the [[#set declib|decay]] and [[#set nfylib|fission yield]] library file paths. | ||
| + | *The calculation requires the [[Defining material volumes|material volumes to be correctly set]]. | ||
| + | *The volume is required for calculating microscopic absorption cross sections matching macroscopic absorption cross sections with poison nuclide densities smeared to the homogenization volume. | ||
| + | *Separate treatment for <sup>135m</sup>Xe requires cross sections for this isotope. | ||
| + | *Parameter ''VOL'' was ''VR'' (optional) in versions before 2.1.32 (ratio of fuel volume to the volume of the homogenized zone). | ||
=== set pop === | === set pop === | ||
| − | + | '''set pop''' ''NPG NGEN NSKIP'' [ ''K0 BTCH NEIG'' ] | |
Sets parameters for simulated neutron population in criticality source mode. Input values: | Sets parameters for simulated neutron population in criticality source mode. Input values: | ||
{| | {| | ||
| − | | < | + | | <tt>''NPG''</tt> |
| − | | number of neutrons per generation | + | | : number of neutrons per generation |
|- | |- | ||
| − | | < | + | | <tt>''NGEN''</tt> |
| − | | number of active generations | + | | : number of active generations |
|- | |- | ||
| − | | < | + | | <tt>''NSKIP''</tt> |
| − | | number of inactive generations | + | | : number of inactive generations |
|- | |- | ||
| − | | < | + | | <tt>''K0''</tt> |
| − | | initial guess for k | + | | : initial guess for ''k''<sub>eff</sub> |
|- | |- | ||
| − | | < | + | | <tt>''BTCH''</tt> |
| − | | batching interval | + | | : batching interval (default value is 1 generation per batch) |
| + | |- | ||
| + | | <tt>''NEIG''</tt> | ||
| + | | : number of independent parallel eigenvalue calculations (default is 1) | ||
|} | |} | ||
| − | The simulation is first run for a number of inactive generations to allow the fission source to converge. This is followed by a number of active generations, during which the results are collected. The statistics are divided in batches, and by default each generation forms its own batch. | + | <u>Notes:</u> |
| + | *The simulation is first run for a number of inactive generations to allow the fission source to converge. This is followed by a number of active generations, during which the results are collected. The statistics are divided in batches, and by default each generation forms its own batch. | ||
| + | *Using the pop card sets the mode to criticality source simulation. External source simulation is invoked using [[#set nps|set nps]]. (The two cards are mutually exclusive). | ||
| + | *Convergence of fission source can be monitored using Shannon entropy (input parameters [[#set his|set his]] and [[#set entr|set entr]]). | ||
| + | *Initial guess for ''k''<sub>eff</sub> is 1.0 by default. Setting the value manually may get the simulation going if it terminates on the first generation because of poor initial guess. The value does not affect fission source convergence. | ||
| + | *See detailed descriptions on [[fission source convergence]] and [[statistical effects of batching]]. | ||
| + | *The number of neutrons per generation also affects the memory usage together with [[#set nbuf|set nbuf]]. With a large value, lowering the buffer size might be necessary for the simulation to be runnable. | ||
| − | + | === set powdens === | |
| − | * | + | '''set powdens''' ''PDE'' [ ''MAT'' ] |
| − | * | + | Sets normalization to power density |
| − | *See [[ | + | |
| + | {| | ||
| + | | <tt>''PDE''</tt> | ||
| + | | : power density (in kW/g) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material in which the given power is produced | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *If the material name is omitted, the value corresponds to average power density produced in the system. | ||
| + | *Power density is given in units of kW/g, not W/g used in several other programs. The typical order of magnitude for this parameter in LWR calculations is 20E-3 ... 50E-3. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set flux|set flux]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set power === | ||
| + | |||
| + | '''set power''' ''P'' [ ''MAT'' ] | ||
| + | Sets normalization to total fission power | ||
| + | |||
| + | {| | ||
| + | | <tt>''P''</tt> | ||
| + | | : fission power (in W) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material in which the given power is produced | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *If the material name is omitted, the value corresponds to total fission power produced in the system. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set powdens|set powdens]], [[#set flux|set flux]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set ppid === | ||
| + | |||
| + | '''set ppid''' ''PID'' | ||
| + | Defines the external code process identifier (PID) number to be used for communication in the POSIX-based coupled calculation communications. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''PID''</tt> | ||
| + | | : Process identifier (PID) of the (parent) process that Serpent should communicate with. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Setting up a communication mode will enable the coupled calculation mode. | ||
| + | *<tt>''PID''</tt> 2<sup>22</sup> theoretical maximum for 64-bit system | ||
| + | *The communication options [[#set comfile|set comfile]], [[#set ppid|set ppid]] and [[#set pport|set pport]] are mutually exclusive, aka, multiple signalling modes are not allowed. | ||
| + | *For more information see: [[Coupled_multi-physics_calculations#External_coupling|External coupling]] | ||
| + | |||
| + | === set pport === | ||
| + | |||
| + | '''set pport''' ''PORT'' | ||
| + | Defines the external code port number to be used for communication in the SOCKET-based coupled calculation communications. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''PORT''</tt> | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Setting up a communication mode will enable the coupled calculation mode. | ||
| + | *The communication options [[#set comfile|set comfile]], [[#set ppid|set ppid]] and [[#set pport|set pport]] are mutually exclusive, aka, multiple signalling modes are not allowed. | ||
| + | *For more information see: [[Coupled_multi-physics_calculations#External_coupling|External coupling]] | ||
| + | |||
| + | === set ppw === | ||
| + | |||
| + | '''set ppw''' ''UNI'' ''LAT'' | ||
| + | |||
| + | Turns on the calculation of pin powers and pin power form factors. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : The universe where the pin power distribution is calculated. | ||
| + | |- | ||
| + | | <tt>''LAT''</tt> | ||
| + | | : The lattice where the calculation is performed. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *See separate description of [[Output parameters#Pin-power form factors|output parameters]] in the <tt>[input]_res.m</tt>. | ||
| + | *The form factors can be printed to the [[Automated_burnup_sequence#Output|group constant output]] with [[Input syntax manual#set_coefpara|set coefpara]]. | ||
| + | *Calculation of pin power form factors also requires calculation of assembly discontinuity factors using [[Input syntax manual#set_adf|set adf]]. | ||
| + | |||
| + | === set precsrcf === | ||
| + | |||
| + | '''set precsrcf''' ''FACTOR'' | ||
| + | |||
| + | Set number of pointwise precursors to hold in memory in [[transient simulations]]. A multiplicative factor for the number of neutrons per batch, i.e. setting ''<tt>FACTOR</tt>'' to 10 when running 1000 neutrons per batch will keep 10000 pointwise precursors in memory. | ||
| + | |||
| + | {| | ||
| + | | <tt>''FACTOR''</tt> | ||
| + | | : The factor to multiply the number of neutrons per batch to obtain the number of pointwise precursors to hold in memory. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Default value is 10. | ||
| + | *The number of pointwise precursors held in memory is controlled to the requested number at time interval boundaries. The physical number of precursors is conserved. | ||
| + | *Storing a too low of a number of pointwise precursors can lead to undersampling of certain precursor groups or parts of geometry. | ||
| + | |||
| + | === set precthresh === | ||
| + | |||
| + | '''set precthresh''' ''THRESHOLD'' | ||
| + | |||
| + | Set the weight threshold for creating and storing a new delayed neutron precursor in [[transient simulations]]. | ||
| + | |||
| + | {| | ||
| + | | <tt>''THRESHOLD''</tt> | ||
| + | | : A weight threshold relative to the incoming neutron weight. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Default value is 1. | ||
| + | *If the weight of the delayed neutron precursor would be below the threshold, Russian roulette is played to either increase the weight to the threshold or not store the precursor at all. | ||
| + | *Setting a lower threshold stores more precursors with lower weight whereas a higher threshold stores fewer precursors with higher weight. | ||
| + | |||
| + | === set printelsp === | ||
| + | |||
| + | '''set printelsp''' ''OPT'' | ||
| + | Prints out the stopping power data for electrons/positions generated by the thick-target bremsstrahlung model. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : Option to output the stopping power data from the bremsstrahlung model off (0/no) or on (1/yes), in file <tt>[input]_elsp.m</tt>. The default option is off. | ||
| + | |} | ||
| + | |||
| + | === set printm === | ||
| + | |||
| + | '''set printm''' ''MODE'' [ ''FRAC'' ] | ||
| + | |||
| + | Print material compositions to <tt>[input].bumat[bu]</tt> file during burnup calculation, where "<tt>bu</tt>" is the burnup step. | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : Set printing on (1/yes) or off (0/no). | ||
| + | |- | ||
| + | | <tt>''FRAC''</tt> | ||
| + | | : Optional atomic fraction for printing out decay nuclides (nuclides with no transport cross sections). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Nuclides without transport data are excluded from the output by default. | ||
| + | *A <tt>''FRAC''</tt> value 0.0 will print out all decay nuclides, while 1.0 will not print out any decay nuclides. Value <tt>''FRAC''</tt> will print out nuclides whose atomic fraction in the material is greater than or equal to <tt>''FRAC''</tt>. | ||
| + | |||
| + | === set qparam_dbrc === | ||
| + | |||
| + | '''set qparam_dbrc''' ''QPARAM'' | ||
| + | Sets the Q-parameter (confidence interval) for temperature majorants to be used with Doppler-broadening rejection correction ([[Input syntax manual#set dbrc|set dbrc]] option). Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''QPARAM''</tt> | ||
| + | | : confidence interval | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The parameter should be adjusted when the DBRC majorant cross section is exceeded frequently | ||
| + | *The default value is 2E-5 for the <u>revisited</u> majorant (default method) and reasonable values are within [1E-5, 1E-4] | ||
| + | *The default value is 3.0 for the <u>traditional</u> majorant and reasonable values are within [1.0, 10.0] | ||
| + | |||
| + | === set qparam_tms === | ||
| + | |||
| + | '''set qparam_tms''' ''QPARAM'' | ||
| + | Sets the Q-parameter (confidence interval) for temperature majorants to be used with target motion sampling method ([[Input syntax manual#mat (material definition)|mat]] card). Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''QPARAM''</tt> | ||
| + | | : confidence interval | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The parameter should be adjusted when the TMS majorant cross section is exceeded frequently | ||
| + | *The default value is 2E-5 for the <u>revisited</u> majorant (default method) and reasonable values are within [1E-5, 1E-4] | ||
| + | *The default value is 3.0 for the <u>traditional</u> majorant and reasonable values are within [1.0, 10.0] | ||
| + | |||
| + | === set relfactor === | ||
| + | |||
| + | '''set relfactor''' ''FAC'' | ||
| + | Sets the underrelaxation factor for the [[Coupled multi-physics calculations#Power relaxation|power relaxation]] used in coupled multi-physics calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FAC''</tt> | ||
| + | | : underrelaxation factor | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * Setting the underrelaxation factor to 0, disables power relaxation altogether. The power distribution written to the output-files will be unrelaxed and only based on the most recent iteration. | ||
| + | * Default value is 1.0 | ||
| + | |||
| + | === set repro === | ||
| + | |||
| + | '''set repro''' ''MODE'' | ||
| + | Sets the reproducibility mode in parallel calculations. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : reproducibility mode | ||
| + | |} | ||
| + | |||
| + | The possible settings for mode are: | ||
| + | |||
| + | {| class="wikitable" style="text-align: left;" | ||
| + | ! Mode | ||
| + | ! Description | ||
| + | |- | ||
| + | | <tt>0</tt> | ||
| + | | No reproducibility | ||
| + | |- | ||
| + | | <tt>1</tt> | ||
| + | | Reproducibility with OpenMP parallelization (default value) | ||
| + | |- | ||
| + | | <tt>2</tt> | ||
| + | | Reproducibility with MPI and hybrid OpenMP / MPI parallelization | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The reproducibility in OpenMP parallelization means that the random number sequences are the same in spite of parallelization. This requires that the histories are calculated always in the same order. This is achieved by sorting the fission banks between batches. With large neutron populations per batches the sorting takes a substantial amount of time which might affect the obtained parallel calculation scalability. | ||
| + | *The reproducibility is often a requirement for debugging the program. <tt>''MODE''</tt> 2 should only be used for debugging. | ||
| + | |||
| + | === set rfr === | ||
| + | |||
| + | '''set rfr''' ''STEP'' ''FILE'' [ ''NFILE'' ] | ||
| + | |||
| + | '''set rfr idx''' ''I'' ''FILE'' [ ''NFILE'' ] | ||
| + | |||
| + | Reads material compositions from a binary restart file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''STEP''</tt> | ||
| + | | : burnup step from which the compositions are obtained | ||
| + | |- | ||
| + | | <tt>''I''</tt> | ||
| + | | : burnup step index from which the compositions are obtained | ||
| + | |- | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : name of the binary restart file | ||
| + | |- | ||
| + | | <tt>''NFILE''</tt> | ||
| + | | : number of restart files | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * If it is not specified a burnup or days step explicitly by preceding the depletion step value by '''bu''' or '''days''', respectively, then, positive values for step are interpreted as burnup units (MWd/kgU) and negative values as time units (days). | ||
| + | * The '''continue''' option sets the restart at the latest calculated depletion step. | ||
| + | * This option can be used together with the [[#set rfw|set rfw]] feature for applying changes in the modeled system during burnup calculation. | ||
| + | * The name of the binary restart file is invariant to domain decomposition, [[#set dd|set dd]]. It corresponds with the standard name of the restart file (without the <tt>_dd[MPIID]</tt> suffix). | ||
| + | * This option reads a single restart file except if the domain decomposition feature is in place (reading the multiple/split restart files generated from the domain decomposition calculation). | ||
| + | * If a non-domain decomposition simulation requires reading the multiple/split restart files from a former one, the number of restart files <tt>''NFILE''</tt> should be specified. In any other case, the number of domains/MPI tasks should match between the writing and reading simulations. The material depletion zone divisions between the writing and reading simulations should agree too. | ||
| + | |||
| + | === set rfw === | ||
| + | |||
| + | '''set rfw''' ''OPT'' [ ''FILE'' ] | ||
| + | Writes material compositions in burnup calculation into a binary restart file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch writing on (0 = no, >0 = yes; with, 1= all nuclides, 2 = transport nuclides) | ||
| + | |- | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : name of the binary restart file. Default value <tt>[input].wrk</tt>. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Restart file writing is switched off by default. | ||
| + | *Version 2.2.0 includes the option to select whether to write all nuclides (default behavior in previous versions) or only the transport nuclides. Before <tt>''OPT''</tt> was limited to 0/1. | ||
| + | *This option can be used together with the [[#set rfr|set rfr]] feature for applying changes in the modeled system during burnup calculation | ||
| + | *This option when is used with the domain decomposition feature, [[#set dd|set dd]], generates multiple restart files - as many as domains (MPI tasks) are defined; the files are named adding <tt>_dd[mpiid]</tt> (domain decomposition identifier) to the standard file name. | ||
| + | |||
| + | === set rnddec === | ||
| + | |||
| + | '''set rnddec''' ''DEC'' [ ''FY DH'' ] | ||
| + | Option to set decay and fission yield data randomized mode on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''DEC''</tt> | ||
| + | | : option to switch randomized decay constants on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''FY''</tt> | ||
| + | | : option to switch randomized fission yields on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''DH''</tt> | ||
| + | | : option to switch randomized decay heat on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * The default mode for either of the options is off. | ||
| + | |||
| + | === set root === | ||
| + | |||
| + | '''set root''' ''UNI'' | ||
| + | Sets the root universe. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Root universe is the universe at the lowest level of the geometry hierarchy, and must always be defined. By default the root is set to "0". | ||
| + | *For more information, see detailed description on the [[universe-based geometry type in Serpent]]. | ||
| + | |||
| + | === set roulette === | ||
| + | |||
| + | '''set roulette''' ''W0'' ''P'' | ||
| + | Sets parameters for weight cut-off and Russian roulette. | ||
| + | |||
| + | {| | ||
| + | | <tt>''W0''</tt> | ||
| + | | : minimum particle weight below which cut-off is applied | ||
| + | |- | ||
| + | | <tt>''P''</tt> | ||
| + | | : survival probability for Russian roulette (default value: 0.5) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Weight cut-off is applied after each collision. | ||
| + | *Can be used together with implicit capture, default value: <tt>''P''</tt> = 0.001 (see [[#set impl|set impl]]) | ||
| + | |||
| + | === set runtme === | ||
| + | |||
| + | '''set runtme''' ''T'' | ||
| + | Sets the maximum running time for the transport simulation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''T''</tt> | ||
| + | | : wall-clock running time (minutes) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *When defined, the transport simulation is terminated after the maximum time is reached. | ||
| + | *Setting the parameter does not override the [[#set pop|set pop]] or [[#set nps|set nps]] option. | ||
| + | |||
| + | === set samarium === | ||
| + | |||
| + | '''set samarium''' ''OPT'' [ ''MAT<sub>1</sub> MAT<sub>2</sub>'' ... ] | ||
| + | |||
| + | Sets equilibrium samarium calculation on or off: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set equilibrium samarium calculation on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub> MAT<sub>2</sub> ...''</tt> | ||
| + | | : optional list of materials for which to set the option (on/off). Default is all fissile materials. Setting equilibrium samarium calculation '''off''' for a list of materials sets it '''on''' for all other fissile materials. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The equilibrium concentration is calculated on depletion zone basis. You may want to divide your fuel material into [[Input syntax manual#div|depletion zones]] | ||
| + | *The equilibrium concentration calculation requires the [[Defining material volumes|material volumes to be correctly set]]. | ||
| + | *The equilibrium concentration calculation requires the [[Input syntax manual#set nfylib|fission yield]] and [[Input syntax manual#set declib|decay]] libraries. | ||
| + | *The equilibrium concentration of samarium is updated according to the batching interval set in the [[#set pop|set pop]] card. Having a large batching interval means that the equilibrium concentration may take a large number of cycles to converge. | ||
| + | |||
| + | === set savesrc === | ||
| + | |||
| + | '''set savesrc''' ''PATH'' [ ''PN PP N<sub>X</sub> N<sub>Y</sub> N<sub>Z</sub>'' ] | ||
| + | |||
| + | Sets up the creation of an initial source to be used in a dynamic simulation with delayed neutron emission. | ||
| + | |||
| + | {| | ||
| + | | <tt>''PATH''</tt> | ||
| + | | : The path of the source file to be created | ||
| + | |- | ||
| + | | <tt>''PN''</tt> | ||
| + | | : Fraction of tentative neutrons to save (default 1.0). Only affects criticality source simulations. | ||
| + | |- | ||
| + | | <tt>''PP''</tt> | ||
| + | | : Fraction of tentative precursors to save (default 1.0). Only affects criticality source simulations. | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : Precursor mesh size in x-direction (default 1) | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : Precursor mesh size in y-direction (default 1) | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : Precursor mesh size in z-direction (default 1) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Four source files will be generated <tt>[''PATH''].main</tt>, <tt>[''PATH''].prec</tt>, <tt>[''PATH''].live</tt> and <tt>[''PATH''].precpoints</tt> | ||
| + | *If used in criticality source simulation, the system should be critical and the values will correspond to steady state values. | ||
| + | *If used in a dynamic simulation, the values will correspond to end-of-simulation values | ||
| + | *Before version 2.2.0, if you are getting a warning from function WriteSourceFile "P larger than 1", you should lower the <tt>''PN''</tt> value. | ||
| + | *See [[Transient simulations]]. | ||
| + | |||
| + | === set sca === | ||
| + | |||
| + | '''set sca''' ''NI'' ''NB'' ''MSH'' | ||
| + | ''MIN<sub>1</sub>'' ''MAX<sub>1</sub>'' ''SZ<sub>1</sub>'' | ||
| + | ''MIN<sub>2</sub>'' ''MAX<sub>2</sub>'' ''SZ<sub>2</sub>'' | ||
| + | ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ''SZ<sub>3</sub>'' | ||
| + | [ ''SUB<sub>1</sub>'' ''SUB<sub>2</sub>'' ''SUB<sub>3</sub>'' ''LIM'' ] | ||
| + | |||
| + | '''set sca''' ''NI'' ''NB'' ''MSH'' | ||
| + | ''X<sub>0</sub>'' ''Y<sub>0</sub>'' | ||
| + | ''P'' ''NX'' ''NY'' | ||
| + | ''MIN<sub>3</sub>'' ''MAX<sub>3</sub>'' ''SZ<sub>3</sub>'' | ||
| + | [ ''SUB<sub>1</sub>'' ''SUB<sub>2</sub>'' ''SUB<sub>3</sub>'' ''LIM'' ] | ||
| + | |||
| + | Invokes a response matrix solver to obtain an improved source guess for criticality source simulations. | ||
| + | |||
| + | {| | ||
| + | | <tt>''NI''</tt> | ||
| + | | : Number of outer iterations | ||
| + | |- | ||
| + | | <tt>''NB''</tt> | ||
| + | | : Number of source batches to collect results | ||
| + | |- | ||
| + | | <tt>''MSH''</tt> | ||
| + | | : mesh type (1 = Cartesian, 2 = Cylindrical, 4 = x-type hexagonal, 5 = y-type hexagonal) | ||
| + | |- | ||
| + | | <tt>''MIN<sub>n</sub>''</tt> | ||
| + | | : minimum mesh boundary (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''MAX<sub>n</sub>''</tt> | ||
| + | | : maximum mesh boundary (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''SZ<sub>n</sub>''</tt> | ||
| + | | : number of mesh cells (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>'', ''Y<sub>0</sub>''</tt> | ||
| + | | : mesh center of hexagonal mesh (currently must be centered at the origin) | ||
| + | |- | ||
| + | | <tt>''P''</tt> | ||
| + | | : hexagonal cell pitch | ||
| + | |- | ||
| + | | <tt>''NX'', ''NY''</tt> | ||
| + | | : hexagonal mesh size | ||
| + | |- | ||
| + | | <tt>''SUB<sub>n</sub>''</tt> | ||
| + | | : number of sub-mesh cells (''n''th coordinate) | ||
| + | |- | ||
| + | | <tt>''LIM''</tt> | ||
| + | | : convergence criterion (typical value 1E-12) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The solver is used to accelerate fission source convergence. | ||
| + | *Cartesian and cylindrical mesh are defined by outer mesh boundaries and number of mesh cells. | ||
| + | *The coordinate axes 1, 2 and 3 in Cartesian mesh refer to (x,y,z) and in cylindrical mesh to (r,θ,z), with θ given in degrees. | ||
| + | *The hexagonal mesh is defined by mesh center, cell pitch, number of cells in the radial dimensions (similar to the hexagonal lattice) and axial binning. | ||
| + | *The mesh must be defined slightly larger than the geometry (the mesh boundaries should not coincide with the geometry boundaries). | ||
| + | *Sub-division is used to improve the solution inside mesh cells. | ||
| + | *The methodology is described in a related article.<ref>Leppänen, J. ''"Acceleration of fission source convergence in the Serpent 2 Monte Carlo code using a response matrix based solution for the initial source distribution."'' Ann. Nucl. Energy [https://doi.org/10.1016/j.anucene.2018.12.044 '''128''' (2019) 63-68]</ref> | ||
| + | *The methodology is <u>still under development</u> and, the input syntax may change in future updates. | ||
| + | |||
| + | === set seed === | ||
| + | |||
| + | '''set seed''' ''RNG'' [ ''NBTCH'' ] | ||
| + | Sets the seed value for the random number sequence. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''RNG''</tt> | ||
| + | | : seed value used for the random number sequence | ||
| + | |- | ||
| + | | <tt>''NBTCH''</tt> | ||
| + | | : starting batch (positive value) or history (negative value) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *By default, Serpent gets the RNG seed from system time. This option overrides the value. | ||
| + | |||
| + | === set sfbuf === | ||
| + | |||
| + | '''set sfbuf''' ''SIZE'' | ||
| + | Sets the size of the source file buffer including, e.g., criticality source, secondary photon source, detector source or dynamic source modes. Input value: | ||
| + | |||
| + | {| | ||
| + | | <tt>''SIZE''</tt> | ||
| + | | : buffer size | ||
| + | |} | ||
| + | |||
| + | === set sfrate === | ||
| + | |||
| + | '''set sfrate''' ''S'' [ ''MAT'' ] | ||
| + | |||
| + | Sets normalization to total spontaneous fission rate. | ||
| + | |||
| + | {| | ||
| + | | <tt>''S''</tt> | ||
| + | | : number of spontaneous fission reactions per second (1/s) | ||
| + | |- | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : dummy parameter | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set power|set power]], [[#set powdens|set powdens]], [[#set flux|set flux]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set srcrate|set srcrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set sfylib === | ||
| + | |||
| + | '''set sfylib''' ''LIB<sub>1</sub>'' [ ''LIB<sub>2</sub>'' ''LIB<sub>3</sub>'' ... ] | ||
| + | Sets the spontaneous fission yield library file paths. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LIB<sub>n</sub>''</tt> | ||
| + | | : library file paths | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Spontaneous fission yield libraries are standard ENDF format files containing spontaneous fission yield data. | ||
| + | *If the file path contains special characters it is advised to enclose it within quotes | ||
| + | |||
| + | === set shbuf === | ||
| + | |||
| + | '''set shbuf''' [ ''OPT_BUF OPT_RES2'' ] | ||
| + | |||
| + | Option to use a shared or private scoring buffer or results array. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT_BUF''</tt> | ||
| + | | : use shared (1/yes) or private (0/no) scoring buffer (BUF, default private) | ||
| + | |- | ||
| + | | <tt>''OPT_RES2''</tt> | ||
| + | | : use shared (1/yes) or private (0/no) results array (RES2, default shared) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Serpent stores scores in a temporary buffer, which in OpenMP parallel mode can be either private (each thread writes in its own buffer) or shared (all threads write in same buffer using atomic operations). Using private buffers increases memory usage to some extent, but it should improve scalability since no barriers need to be set to protect memory. | ||
| + | *Memory consumption of shared RES2 is usually larger than that of shared BUF. | ||
| + | |||
| + | === set sie === | ||
| + | |||
| + | '''set sie''' ''ITER'' | ||
| + | Chooses the [[Stochastic Implicit Euler burnup scheme]] to be used for the burnup calcualtion. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''ITER''</tt> | ||
| + | | : tolerance (negative value) or maximum number of iterations (positive value) for each burnup step | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The SIE burnup scheme is mainly intended for burnup calculations that develop instabilities using the default predictor corrector methods. | ||
| + | *The convergence criteria is set either by the tolerance or the maximum number of iterations. If the tolerance is given, <tt>''ITER''</tt> < 0, the maximum number of iterations is set to 500. If the maximum number iterations is given, <tt>''ITER''</tt> > 0, the tolerance is set to 0.0. | ||
| + | |||
| + | === set sourcescale === | ||
| + | |||
| + | '''set sourcescale''' ''NB'' ''SF<sub>1</sub>'' ''SF<sub>2</sub>'' ... | ||
| + | Defines a time-interval dependent source scaling factor in dynamic external source calculations ([[#set dynsrc|set dynsrc]]). Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NB''</tt> | ||
| + | | : number of time intervals | ||
| + | |- | ||
| + | | <tt>''SF<sub>n</sub>''</tt> | ||
| + | | : time-interval-wise source scaling factor | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The number of time-intervals <tt>''NB''</tt> should match the definition of the time-bin structure (see [[#tme|tme card]]) | ||
| + | |||
| + | === set spa === | ||
| + | |||
| + | '''set spa''' ''CMAP'' [ ''FRAC'' ] | ||
| + | Sets parameters for the source point animation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''CMAP''</tt> | ||
| + | | : color map used for plotting the source point animation | ||
| + | |- | ||
| + | | <tt>''FRAC''</tt> | ||
| + | | : fraction for the color scheme (minimum/maximum) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * The color maps are: 1 - hot; 2 - cold; 4 - jet; 5 - black and white; 6 - hsv; 7 - spring; 8 - summer; 9 - autumn; 10 - winter; 11 - green-purple; 12 - purple-orange; 13 - blue-red. Many of these correspond to what is used in Matlab. | ||
| + | * The default fraction for the color scheme is 1.0. | ||
| + | |||
| + | === set spd === | ||
| + | |||
| + | '''set spd''' ''V<sub>n</sub>'' ''V<sub>p</sub>'' | ||
| + | Overrides the speed of simulated particles. Input values: | ||
| + | |||
| + | {| | ||
| + | | ''V<sub>n</sub>'' | ||
| + | | : speed of neutrons (cm/s) | ||
| + | |- | ||
| + | | ''V<sub>p</sub>'' | ||
| + | | : speed of photons (cm/s) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *This option is intended for adjusting particle speeds for better visualization in [[#set tpa|track plot animations]]. | ||
| + | *Adjusting the speed (obviously) results in incorrect estimates for all time constants. | ||
| + | *Exceeding the speed of light causes a fatal error in debug mode. | ||
| + | |||
| + | === set srcrate === | ||
| + | |||
| + | '''set srcrate''' ''S'' | ||
| + | Sets normalization to total source rate. | ||
| + | |||
| + | {| | ||
| + | | <tt>''S''</tt> | ||
| + | | : source rate (in particles per second) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter. | ||
| + | *Neutron transport simulations are by default normalized to unit total loss rate. | ||
| + | *Photon transport simulations are by default normalized to unit total source rate. | ||
| + | *For other normalization options, see: [[#set powdens|set powdens]], [[#set power|set power]], [[#set flux|set flux]], [[#set genrate|set genrate]], [[#set fissrate|set fissrate]], [[#set absrate|set absrate]], [[#set lossrate|set lossrate]], [[#set sfrate|set sfrate]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set stl === | ||
| + | |||
| + | '''set stl''' ''EXD'' ''NLOOP'' | ||
| + | Sets options for a STL geometry model | ||
| + | |||
| + | {| | ||
| + | | <tt>''EXD''</tt> | ||
| + | | : facet uncertainty margin or exclusion distance (default: 1E-05) | ||
| + | |- | ||
| + | | <tt>''NLOOP''</tt> | ||
| + | |: maximum number of trials (default: 1000) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | * Delta-tracking is enforced by default in STL geometries. | ||
| + | |||
| + | === set stlfile === | ||
| + | |||
| + | '''set stlfile''' ''OPT'' | ||
| + | Sets option to read and write STL search mesh into a binary file, <tt>[input].smh</tt> | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch reading/writing on (1/yes) or off (0/no) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *STL mesh search file reading/writing is switched off by default. | ||
=== set tcut === | === set tcut === | ||
| − | + | '''set tcut''' ''T<sub>max</sub>'' | |
Sets time cut-off for neutrons and photons. Input values: | Sets time cut-off for neutrons and photons. Input values: | ||
{| | {| | ||
| − | | < | + | | <tt>''T<sub>max</sub>''</tt> |
| − | | time limit for simulated particle histories (in seconds) | + | | : time limit for simulated particle histories (in seconds) |
|} | |} | ||
| − | The time cut-off can be used in both neutron and photon external source simulations, to limit the length of particle histories. | + | <u>Notes:</u> |
| + | *The time cut-off can be used in both neutron and photon external source simulations, to limit the length of particle histories. | ||
| + | *Time or generation cut-off ([[#set gcut|set gcut]]) is always needed for neutron external source simulations in super-critical systems. | ||
| + | *Time cut-off is automatically set in the [[dynamic external source simulation mode]]. | ||
| + | *''Note to developers: this should take independent values for photons and neutrons'' | ||
| − | + | === set title === | |
| − | * | + | '''set title''' ''NAME'' |
| − | * | + | Sets a title for the calculation. Input values: |
| + | |||
| + | {| | ||
| + | | <tt>''NAME''</tt> | ||
| + | | : title used for the calculation | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The title is printed in the run-time output. If the title is not set, the input file name is printed instead. | ||
| + | |||
| + | === set tpa === | ||
| + | |||
| + | '''set tpa''' ''T<sub>min</sub>'' ''T<sub>max</sub>'' ''TAIL'' ''NF'' ''EVB'' | ||
| + | Sets parameters for track plot animation. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''T<sub>min</sub>''</tt> | ||
| + | | : starting time of track plot animation (in seconds) | ||
| + | |- | ||
| + | | <tt>''T<sub>max</sub>''</tt> | ||
| + | | : end time of track plot animation (in seconds) | ||
| + | |- | ||
| + | | <tt>''TAIL''</tt> | ||
| + | | : tail length of plotted particles (in cm) | ||
| + | |- | ||
| + | | <tt>''NF''</tt> | ||
| + | | : number of frames | ||
| + | |- | ||
| + | | <tt>''EVB''</tt> | ||
| + | | : event bank size | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The track plot animation works with the [[#plot (geometry plot definition)|geometry plotter]] by creating a number of frames that visualize the motion of particles through the geometry. | ||
| + | *The track plotter is invoked using the "-tracks ''N''" command line option, where ''N'' is the number of simulated particle histories (if the '''set tpa''' option is not defined, the code plots particle tracks in a geometry plot output). | ||
| + | *The routine produces ''NF'' frames <tt>[input]_trck[n]_frame[m].png</tt>, where <tt>[input]</tt> is the input file name, <tt>[n]</tt> is an index corresponding to the geometry plot and <tt>[m]</tt> is the frame index. The frames can be converted into a gif animation using tools like [http://www.imagemagick.org Imagemagick]. | ||
| + | *The plotted particles have a tail to better visualize their movement from one collision to the next. The length of this tail is given in centimeters (similar to geometry dimensions). Neutrons are plotted in magenta, photons are plotted in green. | ||
| + | *Storing the simulated collision points requires additional memory, determined by the event bank size. If the given size is insufficient the calculation is terminated with an error message ("Event bank is empty"). | ||
| + | *Producing track plot animations may require adjusting the particle buffers ([[#set nbuf|set nbuf]] and [[#set gbuf|set gbuf]]). | ||
| + | *The calculation may require a lot of memory for storing the complete simulated histories in neutron-multiplying systems or when particles are split by variance reduction. | ||
| + | *Motion of thermal and fast neutrons cannot be captured simultaneously because of the several orders of magnitude difference in their speed. Thermal systems are best visualized by enforcing all neutrons to travel at the same speed using the [[#set spd|set spd]] option. | ||
| + | *See also [[Visualizing the results#Track plots|detailed description]] on track plotter and track plot animation. | ||
| + | |||
| + | === set transmurea === | ||
| + | |||
| + | '''set transmurea''' ''ZAI<sub>1</sub>'' ''MT<sub>1</sub>'' ''ZAI<sub>2</sub>'' ''MT<sub>2</sub>'' ... | ||
| + | Overrides the automatically generated transmutation paths with a user-provided list of reactions. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''ZAI<sub>n</sub>''</tt> | ||
| + | | : nuclide identifier. | ||
| + | |- | ||
| + | | <tt>''MT<sub>n</sub>''</tt> | ||
| + | | : reaction identifier ([[ENDF_reaction_MT%27s_and_macroscopic_reaction_numbers#ENDF_Reaction_MTs|ENDF reaction MT]]). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The transmutation chains in burnup calculation are by default generated automatically. This option overrides the behavior by including only the reactions included on the list. | ||
| + | *The reactions are given as nuclide identifier - reaction identifier pairs, for example, the radiative capture cross section of <sup>238</sup>U would be 922380 102. | ||
| + | *The nuclide ID can be replaced with "all", in which case all reactions of the type are included. | ||
| + | |||
| + | === set transnorm === | ||
| + | |||
| + | '''set transnorm''' ''FACTOR'' | ||
| + | Multiplies the normalization from a transient source linked with [[Input syntax manual#set dynsrc|set dynsrc]] by a factor. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FACTOR''</tt> | ||
| + | | : factor to multiply the normalization with. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The normalization in transient simulations is, by default, same as in the transient source generation calculation. | ||
| + | *See separate description of [[Transient simulations|transient simulations]]. | ||
| + | |||
| + | === set transtime === | ||
| + | |||
| + | '''set transtime''' ''FLAG'' | ||
| + | Choose how [[#transv|time dependent transformations]] are actually evaluated. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FLAG''</tt> | ||
| + | | : Possible values 0/1: | ||
| + | |- | ||
| + | | | ||
| + | | With '''0''', time dependent transformations are evaluated using the beginning time of the time-interval being simulated (geometry static for each time interval). | ||
| + | |- | ||
| + | | | ||
| + | | With '''1''', time dependent transformations are evaluated using the time when the tracking of the neutron being simulated began (geometry static for each neutron lifetime). | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The default option is 0 (geometry static for each time interval). | ||
| + | *Option 1 generally results in a much smoother transformation. | ||
| + | |||
| + | === set trc === | ||
| + | |||
| + | '''set trc''' ''MAT'' ''FILE'' ''E''<sub>min</sub> [ ''ZAI<sub>1</sub> ZAI<sub>2</sub> ...'' ] | ||
| + | Defines transport correction for the calculation of transport cross section and diffusion coefficient. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MAT''</tt> | ||
| + | | : material to which the correction is applied | ||
| + | |- | ||
| + | | <tt>''FILE''</tt> | ||
| + | | : file path to correction curve data | ||
| + | |- | ||
| + | | <tt>''E''<sub>min</sub></tt> | ||
| + | | : minimum energy above which the correction is applied | ||
| + | |- | ||
| + | | <tt>''ZAI<sub>n</sub>''</tt> | ||
| + | | : nuclide identifiers ([[Definitions, units and constants#definitions|ZAI]]) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *The method calculates transport cross sections by multiplying material total cross sections by a user-defined energy-dependent transport correction ratio before collapsing the energy variable. These transport cross sections are used for calculating diffusion coefficients as <math>D = 1/3\Sigma_\mathrm{tr}</math>. | ||
| + | *If the correction ratios are properly defined, the transport cross section is equivalent with the in-scattering approximation. | ||
| + | *The out-scattering approximation of transport cross section is used for materials without defined correction ratios and energies below the given minimum energy. | ||
| + | *The produced homogenized cross sections are written in variables TRC_TRANSPXS and TRC_DIFFCOEF in the [[Output parameters|<tt>[input]_res.m</tt>]] and [[Automated burnup sequence#Output|<tt>[input].coe</tt>]] output files. | ||
| + | *See separate description of correction factor [[Energy-dependent transport correction factor|file format]]. | ||
| + | *If nuclide identifiers are given the transport correction is applied only to the total cross sections of these nuclides in the given material. In this case the correction should also be given as the ratio of the sum of transport cross sections of these nuclides and the sum of total cross sections of these nuclides. Otherwise the correction is applied to the total cross section of the material. | ||
| + | *Method can not be used for divided or burnable materials for the time being. | ||
| + | |||
| + | === set ttacut === | ||
| + | |||
| + | '''set ttacut''' ''TTACUT'' | ||
| + | |||
| + | Sets cut-off for linear chains method (TTA) based on passage. The cut-off is set to 1E-28 by default. Setting <tt>''TTACUT''</tt> cut-off to a higher value serves as a termination criterion where any chain accounting for less than the fraction <tt>''TTACUT''</tt> of the total atomic density is ignored. | ||
| + | |||
| + | === set ttb === | ||
| + | |||
| + | '''set ttb''' ''OPT'' | ||
| + | Sets the thick-target bremsstrahlung approximation for modelling electrons and positrons on and off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set thick-target bremsstrahlung approximation off (0/no) or on (1/yes). The default option is on. | ||
| + | |} | ||
| + | |||
| + | === set ttbpm === | ||
| + | |||
| + | '''set ttbpm''' ''OPT'' | ||
| + | Sets a separate bremsstrahlung model for positrons. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set a separate bremsstrahlung model off (0/no) or on (1/yes). The default option is on. | ||
| + | |} | ||
| + | |||
| + | === set ufs === | ||
| + | |||
| + | '''set ufs''' ''MODE ORDER N<sub>X</sub> N<sub>Y</sub> N<sub>Z</sub>'' | ||
| + | |||
| + | '''set ufs''' ''MODE ORDER LAT N<sub>Z</sub> Z<sub>MIN</sub> Z<sub>MAX</sub>'' | ||
| + | |||
| + | '''set ufs''' ''MODE ORDER N<sub>X</sub> X<sub>MIN</sub> X<sub>MAX</sub> N<sub>Y</sub> X<sub>MIN</sub> X<sub>MAX</sub> N<sub>Z</sub> Z<sub>MIN</sub> Z<sub>MAX</sub>'' | ||
| + | |||
| + | Turns on the uniform fission source method. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : reaction rate for the weighted mesh distribution (1 = total (collision), 2 = flux, 3 = fission) | ||
| + | |- | ||
| + | | <tt>''ORDER''</tt> | ||
| + | | : exponential factor to adjust the steepness of the distribution | ||
| + | |- | ||
| + | | <tt>''N<sub>X</sub>''</tt> | ||
| + | | : number of x-mesh cells (type 1 and 3) | ||
| + | |- | ||
| + | | <tt>''X<sub>MIN</sub>''</tt> | ||
| + | | : minimum x-coordinate mesh boundary (type 3) | ||
| + | |- | ||
| + | | <tt>''X<sub>MAX</sub>''</tt> | ||
| + | | : maximum x-coordinate mesh boundary (type 3) | ||
| + | |- | ||
| + | | <tt>''N<sub>Y</sub>''</tt> | ||
| + | | : number of y-mesh cells (type 1 and 3) | ||
| + | |- | ||
| + | | <tt>''Y<sub>MIN</sub>''</tt> | ||
| + | | : minimum y-coordinate mesh boundary (type 3) | ||
| + | |- | ||
| + | | <tt>''Y<sub>MAX</sub>''</tt> | ||
| + | | : maximum y-coordinate mesh boundary (type 3) | ||
| + | |- | ||
| + | | <tt>''N<sub>Z</sub>''</tt> | ||
| + | | : number of z-mesh cells (type 1, 2 and 3) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MIN</sub>''</tt> | ||
| + | | : minimum z-coordinate mesh boundary (type 2 and 3) | ||
| + | |- | ||
| + | | <tt>''Z<sub>MAX</sub>''</tt> | ||
| + | | : maximum z-coordinate mesh boundary (type 2 and 3) | ||
| + | |- | ||
| + | | <tt>''LAT''</tt> | ||
| + | | : lattice name defining the mesh structure (type 2) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The UFS method collects the reaction rate distribution during inactive neutron cycles and adjust the number of emitted fission neutrons during the active cycles by a factor for a given mesh cell <tt>''i''</tt>: <math>\overline{f_j^r} / f_i^r</math> for <tt>''f''</tt> the distribution and <tt>''r''</tt> the exponential factor. | ||
| + | *The mesh definition can cover the whole geometry (type 1 definition, 3 parameters), be fixed to a lattice (type 2 definition, 4 parameters) or delimited within a region by the number of mesh cells and boundaries in x-, y- and z- directions (type 3 definition, 9 parameters). | ||
| + | * The lattice definition fixes the mesh structure in x- and y- directions. The lattice can be 2D square or hexagonal type defined in the global coordinate system. | ||
| + | *Defining the exponential factor <tt>''ORDER''</tt> to 1.0 the method results in a relatively uniform distribution of source points. | ||
| + | *An additional mode, <tt>''MODE''</tt> '''4''', has been set to use the uniform fission method coupled with the built-in response matrix solver. The mesh definition is carried out by the response matrix mesh, see see [[#set sca|set sca]]. (The exponential factor is set to 1.0). | ||
| + | *See Serpent forum topic on [http://ttuki.vtt.fi/serpent/viewtopic.php?f=24&t=1762 UFS method]. | ||
| + | |||
| + | === set ures === | ||
| + | |||
| + | '''set ures''' ''OPT'' [ ''NUC<sub>1</sub> NUC<sub>2</sub> ... '' ][ ''DILUCUT'' ] | ||
| + | Sets unresolved resonance probability table sampling on or off. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch probability table sampling on (1/yes) or off (0/no) | ||
| + | |- | ||
| + | | <tt>''NUC<sub>n</sub>''</tt> | ||
| + | | : list of nuclides to which the option is applied to, e.g. "92238.09c". Setting unresolved resonance probability table sampling '''off''' for a list of nuclides will set it '''on''' for the rest of the nuclides. | ||
| + | |- | ||
| + | |<tt>''DILUCUT''</tt> | ||
| + | | : infinite dilution cut-off | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Probability table sampling is switched off by default | ||
| + | *The infinite dilution cut-off <tt>''DILUCUT''</tt> defines a limit for atomic fractions, and probability table sampling is used only for nuclides with concentration above this limit. The cut-off is set to 1E-09 by default. | ||
| + | *See separate description of [[physics options in Serpent]] for differences to other codes. | ||
| + | |||
| + | === set usym === | ||
| + | |||
| + | '''set usym''' ''UNI AX BC X<sub>0</sub> Y<sub>0</sub> θ<sub>0</sub> θ<sub>w</sub>'' [ ''OPT'' ] | ||
| + | |||
| + | Defines a universe symmetry. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''UNI''</tt> | ||
| + | | : universe name | ||
| + | |- | ||
| + | | <tt>''AX''</tt> | ||
| + | | : symmetry axis (1 = x, 2 = y, 3 = z) | ||
| + | |- | ||
| + | | <tt>''BC''</tt> | ||
| + | | : boundary condition (2 = reflective, 3 = periodic) | ||
| + | |- | ||
| + | | <tt>''X<sub>0</sub>''</tt> | ||
| + | | : x-coordinate of the origin | ||
| + | |- | ||
| + | | <tt>''Y<sub>0</sub>''</tt> | ||
| + | | : y-coordinate of the origin | ||
| + | |- | ||
| + | | <tt>''θ<sub>0</sub>''</tt> | ||
| + | | : azimuthal position where the symmetry segment starts (degrees) | ||
| + | |- | ||
| + | | <tt>''θ<sub>w</sub>''</tt> | ||
| + | | : width of the segment (degrees) | ||
| + | |- | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to use actual reflections and translations instead of coordinate transformations | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *Universe symmetries can be used to simplify construction of complex geometries. | ||
| + | *Symmetries can also be used to reduce the number of burnable material zones when [[automated depletion zone division]] is applied. | ||
| + | *When symmetries are used, it is important to pay attention to the [[Defining material volumes|definition of material volumes]]. | ||
| + | *When using symmetries together with lattices, the lattice positions overlapped by the symmetry should remain empty of fuel pins for example to prevent excess memory usage during depletion calculations. | ||
| + | *Universe symmetries are applied by default using coordinate transformations. This means that the particle position and direction are not affected during tracking. This behavior can be overridden by setting the last option to 1. | ||
| + | *The values of surface current and flux [[#det|detectors]] and values produced by for example by [[#set adf|set adf]] cards might not be correct when using actual reflections and transformations. | ||
| + | *Track plots do not work properly with actual reflections and translations. | ||
| + | *For more information, see examples on [[universe symmetries]]. | ||
| + | *The supported Serpent 1 syntax-style: '''set usym''' <tt>''UNI''</tt> <tt>''SYM''</tt> [ <tt>''X<sub>0</sub>''</tt> <tt>''Y<sub>0</sub>''</tt> ] allows only quadrant symmetries (<tt>''SYM''</tt> = 4) in universe 0 (<tt>''UNI''</tt> = 0) centred in origin (<tt>''X<sub>0</sub>''</tt>, <tt>''Y<sub>0</sub>''</tt>) = (0,0). | ||
| + | |||
| + | === set U235H === | ||
| + | |||
| + | '''set U235H''' ''U235_FISSE'' | ||
| + | |||
| + | Sets the U-235 fission heating value. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''U235_FISSE''</tt> | ||
| + | | : fission heating value of U-235 (MeV) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *By default the U-235 fission heating value is set to 202.27 MeV and the values for other actinides scaled based the Q-values found in the cross section libraries. | ||
| + | *See also [[Input_syntax_manual#set_fissh|set fissh]]. | ||
| + | *See also Section 5.8 of [http://montecarlo.vtt.fi/download/Serpent_manual.pdf Serpent 1 User Manual]. | ||
| + | |||
| + | === set voidc === | ||
| + | |||
| + | '''set voidc''' ''OPT'' | ||
| + | Option that enables removing void cells to accelerate particle tracking in complex geometries with numerous void. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch the removal mode on (1/yes) or off (0/no). Default is off. | ||
| + | |} | ||
| + | |||
| + | === set wrnout === | ||
| + | |||
| + | '''set wrnout''' ''OPT'' | ||
| + | Option that enables the generation of a warning-message output file. | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to switch the removal mode on (1/yes) or off (0/no). Default is ON. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The warning-message output file <tt>[input].wrn</tt> collects, as a replica, the errors/warnings/notes printed out in the main (screen) output. | ||
| + | |||
| + | === set wie === | ||
| + | |||
| + | '''set wie''' ''FRAC_ITER'' | ||
| + | Option that enables the Wielandt’s method to accelerate the convergence of the fission source by setting the initial fraction iteration condition. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''FRAC_ITER''</tt> | ||
| + | | : initial guess of the probability of banking the neutron (negative value, ]-1, 0[) or the shifted eigenvalue (positive value, [0.5, 3]) | ||
| + | |} | ||
| + | |||
| + | === set wwb === | ||
| + | |||
| + | '''set wwb''' ''LO'' ''UP'' [ ''F'' ] | ||
| + | |||
| + | Defines the relation between importances and weight-window boundaries. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''LO''</tt> | ||
| + | | : factor relating the lower weight-window boundary to importance | ||
| + | |- | ||
| + | | <tt>''UP''</tt> | ||
| + | | : factor relating the upper weight-window boundary to importance | ||
| + | |- | ||
| + | | <tt>''F''</tt> | ||
| + | | : Russian roulette survival probability factor | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *Weight window boundaries are inversely proportional to importance. These factors define the coefficients. | ||
| + | *Default values are <tt>''LO''</tt> = 0.5, <tt>''UP''</tt> = 2.0, <tt>''F''</tt> = 1.0. | ||
| + | *See also [[Input_syntax_manual#wwin (weight window mesh definition)|weight window mesh definition]]. | ||
| + | |||
| + | === set xenon === | ||
| + | |||
| + | '''set xenon''' ''OPT'' [ ''MAT<sub>1</sub> MAT<sub>2</sub> ...'' ] | ||
| + | |||
| + | Sets equilibrium xenon calculation on or off: | ||
| + | |||
| + | {| | ||
| + | | <tt>''OPT''</tt> | ||
| + | | : option to set equilibrium xenon calculation on or off (0 = off/no, >0 = on/yes; with 1= include only <sup>135</sup>Xe, 2 = include <sup>135</sup>Xe and <sup>135m</sup>Xe) | ||
| + | |- | ||
| + | | <tt>''MAT<sub>1</sub> MAT<sub>2</sub> ...''</tt> | ||
| + | | : optional list of materials for which to set the option (on/off). Default is all fissile materials. Setting equilibrium xenon calculation '''off''' for a list of materials sets it '''on''' for all other fissile materials. | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | *The equilibrium xenon concentration calculation forces the xenon number density to be in equilibrium with the current flux and xenon absorption level during the transport calculation. | ||
| + | *The equilibrium xenon concentration calculation is meant for example to stabilize the [[#dep|burnup calculations]]. | ||
| + | *The equilibrium concentration is calculated on depletion zone basis. You may want to divide your fuel material into [[Input syntax manual#div|depletion zones]] | ||
| + | *The equilibrium concentration calculation requires the [[Defining material volumes|material volumes to be correctly set]]. | ||
| + | *The equilibrium concentration calculation requires the [[Input syntax manual#set nfylib|fission yield]] and [[Input syntax manual#set declib|decay]] libraries. | ||
| + | *The equilibrium concentration of xenon is updated according to the batching interval set in the [[#set pop|set pop]] card. Having a large batching interval means that the equilibrium concentration may take a large number of cycles to converge. | ||
| + | |||
| + | === set xscalc === | ||
| + | |||
| + | '''set xscalc''' ''MODE'' | ||
| + | |||
| + | Calculation mode for transmutation cross sections. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''MODE''</tt> | ||
| + | | : Calculation mode (1 = direct tallies, 2 = spectrum-collapse) | ||
| + | |} | ||
| + | |||
| + | <u>Notes:</u> | ||
| + | |||
| + | *This parameter controls the way transmutation cross sections are calculated in burnup mode. In spectrum-collapse mode these cross sections are calculated after the transport simulation, using a fine-group spectrum collected for each material. | ||
| + | *The spectrum-collapse mode leads to improved performance, but also increased memory footprint per depletion zone. | ||
| + | *The option is automatically set when using optimization modes, and it is not recommended to be defined manually. | ||
| + | *Many of the old example input files set spectrum collapse method on, which overrides the behaviour in lower optimization modes. | ||
| + | *See also [[#set opti|set opti]]. | ||
| + | |||
| + | === set xsplot === | ||
| + | |||
| + | '''set xsplot''' ''NP E<sub>min</sub> E<sub>max</sub>'' | ||
| + | |||
| + | Prints cross section data to <tt>[input]_xs0.m</tt> file. Input values: | ||
| + | |||
| + | {| | ||
| + | | <tt>''NP''</tt> | ||
| + | | : Number of energy points to print (minimum value: 10). | ||
| + | |- | ||
| + | | <tt>''E<sub>min</sub>''</tt> | ||
| + | | : Lower boundary for the energy points. | ||
| + | |- | ||
| + | | <tt>''E<sub>max</sub>''</tt> | ||
| + | | : Upper boundary for the energy points. | ||
| + | |} | ||
| + | |||
| + | The cross sections will be printed out at <tt>''NP''</tt> logarithmically spaced points between the energy boundaries. | ||
== References == | == References == | ||
<references/> | <references/> | ||
| + | |||
| + | [[Category:Input|*]] | ||
Revision as of 21:16, 25 May 2022
Serpent has no interactive user interface. All communication between the code and the user is handled through one or several input files and various output files.
The format of the input file is unrestricted. The file consists of white-space (space, tab or newline) separated words, containing alphanumeric characters(’a-z’, ’A-Z’, ’0-9’, ’.’, ’-’). If special characters or white spaces need to be used within the word (file names, etc.), the entire string must be enclosed within quotes.
The input file is divided into separate data blocks, denoted as cards. The file is processed one card at a time and there are no restrictions regarding the order in which the cards should be organized. The input cards are listed below. Additional options are followed by key word "set". All input cards and options are case-insensitive (note to developers: make it so). Each input card is delimited by the beginning of the next card. It is hence important that none of the parameter strings used within the card coincide with the card identifiers.
The percent-sign ('%') is used to define a comment line. Anything from this character to the end of the line is omitted when the input file is read. Unlike Serpent 1, hashtag ('#') can no longer be used to mark comment lines in Serpent 2 input. The alternative is to use C-style comment sections beginning with "/*" and ending with "*/". Everything between these delimiters is omitted, regardless of the number of newlines or special characters.
This page will contain the whole input syntax of Serpent 2, with links to more detailed descriptions where needed. For reference see also the Serpent 1 input manual[1].
Contents
- 1 Input cards
- 1.1 branch (branch definition)
- 1.2 casematrix (casematrix definition)
- 1.3 cell (cell definition)
- 1.4 coef (coefficient matrix definition)
- 1.5 datamesh (general data mesh definition)
- 1.6 dep (depletion history)
- 1.7 det (detector definition)
- 1.8 div (divisor definition)
- 1.9 dtrans (detector mesh transformation)
- 1.10 ene (energy grid definition)
- 1.11 ftrans (fill transformation)
- 1.12 fun (function definition)
- 1.13 hisv (history variation matrix definition)
- 1.14 ifc (interface file)
- 1.15 include (read another input file)
- 1.16 lat (regular lattice definition)
- 1.17 ltrans (lattice transformation)
- 1.18 mat (material definition)
- 1.19 mesh (mesh plot definition)
- 1.20 mflow (material flow definition)
- 1.21 mix (mixture definition)
- 1.22 nest (nested universe definition)
- 1.23 particle (particle geometry definition)
- 1.24 pbed (explicit stochastic (pebble bed) geometry)
- 1.25 phb (pulse-height Gaussian energy broadening definition)
- 1.26 pin (pin geometry definition)
- 1.27 plot (geometry plot definition)
- 1.28 rep (reprocessor definition)
- 1.29 sample (Temperature / density data sample definition)
- 1.30 sens (sensitivity calculation definition)
- 1.31 solid (irregular 3D geometry definition)
- 1.32 src (source definition)
- 1.33 srtrans (source transformation)
- 1.34 strans (surface transformation)
- 1.35 surf (surface definition)
- 1.36 therm and thermstoch (thermal scattering)
- 1.37 tme (time binning definition)
- 1.38 trans (transformations)
- 1.39 transb (burnup transformation)
- 1.40 transv and transa (velocity and acceleration transformations)
- 1.41 umsh (unstructured mesh-based geometry definition)
- 1.42 utrans (universe transformation)
- 1.43 wwgen (response matrix based importance map solver)
- 1.44 wwin (weight window mesh definition)
- 2 Input options
- 2.1 set absrate
- 2.2 set acelib
- 2.3 set adf
- 2.4 set alb
- 2.5 set arr
- 2.6 set ba
- 2.7 set bala
- 2.8 set bc
- 2.9 set blockdt
- 2.10 set bralib
- 2.11 set branchless
- 2.12 set bumode
- 2.13 set bunorm
- 2.14 set ccmaxiter
- 2.15 set ccmaxpop
- 2.16 set cdop
- 2.17 set cea
- 2.18 set cfe
- 2.19 set cmm
- 2.20 set coefpara
- 2.21 set combing
- 2.22 set comfile
- 2.23 set confi
- 2.24 set coverxlib
- 2.25 set covlib
- 2.26 set cpd
- 2.27 set cpop
- 2.28 set csw
- 2.29 set dataout
- 2.30 set dbrc
- 2.31 set dd
- 2.32 set declib
- 2.33 set decomp
- 2.34 set delnu
- 2.35 set depmtx
- 2.36 set depout
- 2.37 set deppara
- 2.38 set depstepbunorm
- 2.39 set dfsol
- 2.40 set dix
- 2.41 set dspec
- 2.42 set dt
- 2.43 set dynccfile
- 2.44 set dynsrc
- 2.45 set ecut
- 2.46 set ecutdens
- 2.47 set ecutmat
- 2.48 set eddi
- 2.49 set edepdel
- 2.50 set edepkcorr
- 2.51 set edepmode
- 2.52 set egrid
- 2.53 set ekn
- 2.54 set elcond
- 2.55 set elgas
- 2.56 set elmee
- 2.57 set elspn
- 2.58 set entr
- 2.59 set fininitfile
- 2.60 set fissh
- 2.61 set fissrate
- 2.62 set fissye
- 2.63 set flux
- 2.64 set fluxlimtrc
- 2.65 set fmtx
- 2.66 set forcedt
- 2.67 set fpcut
- 2.68 set fsp
- 2.69 set fum
- 2.70 set gbuf
- 2.71 set gct
- 2.72 set gcu
- 2.73 set gcut
- 2.74 set genrate
- 2.75 set gpop
- 2.76 set gsw
- 2.77 set his
- 2.78 set ifp
- 2.79 set imp
- 2.80 set impl
- 2.81 set inftrk
- 2.82 set inventory
- 2.83 set isobra
- 2.84 set iter alb
- 2.85 set iter nuc
- 2.86 set keff
- 2.87 set lossrate
- 2.88 set lost
- 2.89 set maxsplit
- 2.90 set mbtch
- 2.91 set mcvol
- 2.92 set mdep
- 2.93 set memfrac
- 2.94 set mfpcut
- 2.95 set mfpcutdens
- 2.96 set mfpcutmat
- 2.97 set micro
- 2.98 set minxs
- 2.99 set multilevelgcu
- 2.100 set mvol
- 2.101 set nbuf
- 2.102 set nfg
- 2.103 set nfylib
- 2.104 set ngamma
- 2.105 set nphys
- 2.106 set nps
- 2.107 set opti
- 2.108 set outp
- 2.109 set pbuf
- 2.110 set pcc
- 2.111 set pdatadir
- 2.112 set poi
- 2.113 set pop
- 2.114 set powdens
- 2.115 set power
- 2.116 set ppid
- 2.117 set pport
- 2.118 set ppw
- 2.119 set precsrcf
- 2.120 set precthresh
- 2.121 set printelsp
- 2.122 set printm
- 2.123 set qparam_dbrc
- 2.124 set qparam_tms
- 2.125 set relfactor
- 2.126 set repro
- 2.127 set rfr
- 2.128 set rfw
- 2.129 set rnddec
- 2.130 set root
- 2.131 set roulette
- 2.132 set runtme
- 2.133 set samarium
- 2.134 set savesrc
- 2.135 set sca
- 2.136 set seed
- 2.137 set sfbuf
- 2.138 set sfrate
- 2.139 set sfylib
- 2.140 set shbuf
- 2.141 set sie
- 2.142 set sourcescale
- 2.143 set spa
- 2.144 set spd
- 2.145 set srcrate
- 2.146 set stl
- 2.147 set stlfile
- 2.148 set tcut
- 2.149 set title
- 2.150 set tpa
- 2.151 set transmurea
- 2.152 set transnorm
- 2.153 set transtime
- 2.154 set trc
- 2.155 set ttacut
- 2.156 set ttb
- 2.157 set ttbpm
- 2.158 set ufs
- 2.159 set ures
- 2.160 set usym
- 2.161 set U235H
- 2.162 set voidc
- 2.163 set wrnout
- 2.164 set wie
- 2.165 set wwb
- 2.166 set xenon
- 2.167 set xscalc
- 2.168 set xsplot
- 3 References
Input cards
NOTE: Serpent command words are in boldface and input parameters entered by the user in CAPITAL ITALIC. Optional input parameters are enclosed in [ square brackets ], and when the number of values is not fixed, the remaining values are marked with three dots (...).
branch (branch definition)
branch NAME [ repm MAT1 MAT2 ]
[ repu UNI1 UNI2 ]
[ stp MAT DENS TEMP THERM1 SABL1 SABH1 THERM2 SABL2 SABH2 ... ]
[ tra TGT TRANS ]
[ xenon OPT ]
[ samarium OPT ]
[ norm NSF ]
[ gcu UNI2 ]
[ reptrc FILE1 FILE2 ]
[ var VNAME VAL ]
Defines the variations invoked for a branch in the automated burnup sequence. Input values:
| NAME | : branch name |
| MAT1 | : name of the replaced material |
| MAT2 | : name of the replacing material |
| UNI1 | : name of the replaced universe |
| UNI2 | : name of the replacing universe |
| MAT | : name of the material for which density and temperature are adjusted |
| DENS | : material density after adjustment (positive entries for atomic, negative entries for mass densities) |
| TEMP | : material temperature after adjustment, or -1 if no adjustment in temperature |
| THERMn | : nth thermal scattering data associated with the material |
| SABLn | : name of the nth S(α, β) library for temperature below the given value |
| SABHn | : name of the nth S(α, β) library for temperature above the given value |
| TGT | : target universe, surface or cell |
| TRANS | : name of the applied transformation |
| OPT | : option for setting poison concentrations (0 = set to zero, 1 = use values from restart file) |
| NSF | : normalization scaling factor |
| FILE1 | : file path of the replaced transport correction curve data |
| FILE2 | : file path of the replacing transport correction curve data |
| VNAME | : variable name |
| VAL | : variable value |
Notes:
- The branch name identifies the branch in the coefficient matrix of the coef card
- The input parameters consist of a number variations, which are invoked when the branch is applied. A single branch card may inclued one or several variations.
- The repm variation can be used to replace one material with another, for example, to change coolant boron concentration.
- The material replacement works as if MAT1 were created using the mat or mix card of MAT2.
- The name of the material present in the geometry will still be MAT1 after the replacement, but the material specification (composition, density, tmp, moder, rgb, etc.) will correspond to MAT2.
- This means that all other input-cards that are linked to a specific material name such as det dm, src sm, set trc and set iter nuc can be linked to the original material (MAT1) and they will automatically apply to whatever material MAT2 replaces MAT1 for the branch calculation.
- The repu variation can be used to replace one universe with another, for example, to replace empty control rod guide tubes with rodded tubes for control rod insertion in 2D geometries.
- The name of the universe present in the geometry will still be UNI1 after the replacement, but the universe contents will correspond to UNI2.
- This means that all other input-cards that are linked to a specific universe name such as det du and src su can be linked to the original universe (UNI1) and they will automatically apply to whatever universe UNI2 replaces UNI1 for the branch calculation.
- The stp variation can be used to change material density and temperature. The adjustment is made using the built-in Doppler-broadening preprocessor routine and tabular interpolation for S(α, β) thermal scattering data.
- The last three parameters of the stp entry are provided only if the material has thermal scattering libraries attached to it (see the therm card).
- The tra variation can be used to move or rotate different parts of the geometry, for example, to adjust the position of control rods in 3D geometries. The name of the transformation refers to the unit (universe, cell or surface) entry in the trans card.
- The xenon and samarium options can be set to enforce the concentrations of fission product poisons Xe-135 and Sm-149 to zero. By default the concentrations are read from the restart file.
- The norm variation can be used to change the normalization. The adjustment is made applying the parameter NSF as a multiplicative scaling factor to the given normalization.
- The gcu variation can be used to replace the universe for group constant generation. This variation is limited to a single-valued GCU-list.
- The reptrc variation can be used to replace a transport correction file with another.
- Variables can be used to pass information into output file, which may be convenient for the post-processing of the data.
- The branch card is used together with the coef card.
- For more information, see detailed description on the automated burnup sequence.
- The replaced material MAT1 of repm variation is also replaced inside mixtures. This means one can not replace a material with a mixture defined with mix card containing the replaced material (for example replacing pure water defined with mat card by a mixture of boron and water defined with a mix card containing the same pure water material).
- The replacing material MAT2 of repm variation can not be included in geometry using other cards than the branch card with the repm variation, version 2.1.30.
- The "sum" option to define the material density as the sum of the constituent nuclide densities is not supported from version 2.2.0 and on.
casematrix (casematrix definition)
casematrix CASE_NAME
NHIS [ HIS_BR1 HIS_BR2 ... HIS_BRNHIS ]
NBU [ BU1 BU2 ... BUNBU ]
NBR1 [ BR1,1 BR1,2 ... BR1,NBR1 ]
NBR2 [ BR2,1 BR2,2 ... BR1,NBR2 ]
...
Defines the casematrix for the automated burnup sequence. Input values:
| CASE_NAME | : name of the casematrix |
| NHIS | : number of history variations |
| HIS_BRk | : name of the kth history variation branch |
| NBU | : number of burnup points |
| BUn | : burnup steps at which the momentary variation branches are invoked |
| NBRm | : number branches in the mth dimension of the burnup matrix |
| BRm,i | : name of the ith branch in the mth dimension |
Notes:
- The casematrix card performs multiple depletions with NHIS (different) historical variations and performs restarts similar as the coef input card.
- The casematrix card creates a multi-dimensional coefficient matrix (of size NBR1 × NBR2 × NBR3 × ... ). The automated burnup sequence performs a restart for each of the listed burnup points, and loops over the branch combinations defined by the coefficient matrix. This is repeated for each different depletion history.
- Positive values in the burnup vector are interpreted as (MWd/kgU), negative values are interpreted as time steps in days.
- The casematrix card is used together with the branch card and -casematrix running option.
- Multiple casematrix cards can be given in a single input file.
- For more information, see detailed description on automated burnup sequence.
cell (cell definition)
cell NAME UNI0 MAT [ SURF1 SURF2 ... ]
Defines a material cell. Input values:
| NAME | : cell name |
| UNI0 | : universe where the cell belongs to |
| MAT | : material that fills the cell |
| SURFn | : surface list |
cell NAME UNI0 fill UNI1 [ SURF1 SURF2 ... ]
Defines a filled cell. Input values:
| NAME | : cell name |
| UNI0 | : universe where the cell belongs to |
| UNI1 | : universe that fills the cell |
| SURFn | : surface list |
cell NAME UNI0 outside [ SURF1 SURF2 ... ]
Defines an outside cell. Input values:
| NAME | : cell name |
| UNI0 | : universe where the cell belongs to |
| SURFn | : surface list |
Notes:
- There are three types of cells: material cells, filled cells and outside cells. Filled cells are identified by providing the key word fill, followed by the universe filling the cell. If the key word is missing, the third entry is interpreted as the material filling the cell. Outside cells are identified by replacing the material name with key word outside.
- Cells defined without surfaces are treated as infinite, from version 2.1.32 on.
- Void cells can be defined by setting the material name to "void"
- When the geometry is set up, the root universe must always be defined. By default the root universe is named "0", and it can be changed with the set root option.
- Outside cells are used to define the part of the geometry that does not belong to the model. When the particle enters an outside cell, boundary conditions are applied. It is important that the geometry model is non-re-entrant (convex) when vacuum boundary conditions are used. Delta-tracking might miss the boundary conditions in a re-entrant (concave) outer surface.
- Outside cells are allowed only in the root universe. It is important that all space outside the model is defined.
- The surface list defines the boundaries of the cell by listing the surface names (as provided in the surface card), together with the operator identifiers (nothing for intersection, ":" for union, "-" for complement and "#" for cell complement).
- Universes are implicitly declared for example by using the UNI0 key words on cell cards as there is no explicit universe input card.
- For more information, see detailed description on the universe-based geometry type in Serpent.
coef (coefficient matrix definition)
coef NBU [ BU1 BU2 ... ]
[ NBR1 BR1,1 BR1,2 ... ]
[ NBR2 BR2,1 BR2,2 ... ]
...
Defines the coefficient matrix for the automated burnup sequence. Input values:
| NBU | : number of burnup points |
| BUn | : burnup steps at which the branches are invoked |
| NBRm | : number branches in the mth dimension of the burnup matrix |
| BRm,i | : name of the ith branch in the mth dimension |
Notes:
- The coef card creates a multi-dimensional coefficient matrix (of size NBR1 × NBR2 × NBR3 × ... ). The automated burnup sequence performs a restart for each of the listed burnup points, and loops over the branch combinations defined by the coefficient matrix.
- Positive values in the burnup vector are interpreted as (MWd/kgU), negative values are interpreted as time steps in days.
- The coef card is used together with the branch card.
- For multiple historical variations or historical conditions defined using a branch card, see the casematrix card.
- For more information, see detailed description on automated burnup sequence.
datamesh (general data mesh definition)
The datamesh card allows the user to define various meshes that can be used for spatial discretization e.g. in detectors.
datamesh NAME 1 USELC NX XMIN XMAX NY YMIN YMAX NZ ZMIN ZMAX
Defines a regular Cartesian mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| NX | : number of cells in the x direction |
| XMIN | : mesh lower x boundary |
| XMAX | : mesh higher x boundary |
| NY | : number of cells in the y direction |
| YMIN | : mesh lower y boundary |
| YMAX | : mesh higher y boundary |
| NZ | : number of cells in the z direction |
| ZMIN | : mesh lower z boundary |
| ZMAX | : mesh higher z boundary |
datamesh NAME 2 USELC NR RMIN RMAX NPHI
Defines a regular 2D cylindrical mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| NR | : number of cells in the radial direction |
| RMIN | : mesh inner radial boundary |
| RMAX | : mesh outer radial boundary |
| NPHI | : number of cells in the polar angle direction |

datamesh NAME 4 USELC X0 Y0 PITCH ZMIN ZMAX NX NY NZ
Defines a regular x-type hexagonal mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| X0 | : mesh horizontal origin x-coordinate |
| Y0 | : mesh horizontal origin y-coordinate |
| PITCH | : mesh horizontal pitch (equal to cell flat-to-flat width) |
| ZMIN | : mesh lower z boundary |
| ZMAX | : mesh higher z boundary |
| NX | : number of cells in the x direction |
| NY | : number of cells in the y direction |
| NZ | : number of cells in the z direction |

datamesh NAME 5 USELC X0 Y0 PITCH ZMIN ZMAX NX NY NZ
Defines a regular y-type hexagonal mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| X0 | : mesh horizontal origin x-coordinate |
| Y0 | : mesh horizontal origin y-coordinate |
| PITCH | : mesh horizontal pitch (equal to cell flat-to-flat width) |
| ZMIN | : mesh lower z boundary |
| ZMAX | : mesh higher z boundary |
| NX | : number of cells in the x direction |
| NY | : number of cells in the y direction |
| NZ | : number of cells in the z direction |
datamesh NAME 6 USELC NX NY NZ X1 ... XNX+1 Y1 ... YNY+1 Z1 ... ZNZ+1
Defines an irregular Cartesian mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| NX | : number of cells in the x direction |
| NY | : number of cells in the y direction |
| NZ | : number of cells in the z direction |
| Xi | : NX + 1 mesh boundaries in the x direction |
| Yi | : NY + 1 mesh boundaries in the y direction |
| Zi | : NZ + 1 mesh boundaries in the z direction |
datamesh NAME 8 USELC NR NPHI R1 ... RNR+1
Defines a radially irregular 2D cylindrical mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| NR | : number of cells in the radial direction |
| NPHI | : number of cells in the polar angle direction |
| Ri | : NR + 1 mesh boundaries in the r direction |
datamesh NAME 9 NLEVEL MESH1 ... MESHNLEVEL
Defines a regular nested mesh that can be linked to detectors, interfaces etc.
| NAME | : mesh name |
| USELC | : use lowest level coordinates (1/yes) instead of global coordinates (0/no) for the mesh search |
| NLEVEL | : number of nested levels in this mesh |
| MESHi | : sub mesh to use at level i |
Notes:
- When Serpent makes the mesh search for a specific collision point it will save the collision mesh cell temporarily so that the cell search is conducted at most once even when scoring multiple estimators using the same mesh.
dep (depletion history)
dep STYPE [ STEP1 STEP2 ... ]
Defines depletion history with steps and activates depletion calculation mode. Input values:
| STYPE | : step type |
| STEPn | : depletion step list |
The possible step types are:
| Type | Description | Quantity | Unit |
|---|---|---|---|
| bustep | Depletion step | Burnup interval | MWd/kgHM |
| butot | Depletion step | Cumulative burnup | MWd/kgHM |
| daystep | Depletion step | Time interval | d |
| daytot | Depletion step | Cumulative time | d |
| decstep | Decay step | Time interval | d |
| dectot | Decay step | Cumulative time | d |
| actstep | Activation step | Time interval | d |
| acttot | Activation step | Cumulative time | d |
Notes:
- Transport cycle is omitted with the decstep and dectot options.
- Transport cycle is run only once with the actstep and acttot options.
dep pro REP_NAME STYPE [ STEP1 STEP2 ... ]
Links a reprocessor to the depletion calculation. Input values:
| REP_NAME | : reprocessor name |
| STYPE | : step type |
| STEPn | : depletion step list |
Notes:
- The reprocessing system or reprocessor controller is defined using the rep card.
dep bra PTR_BRANCH
det (detector definition)
det NAME [ PART ]
[ dr MT MAT ]
[ dv VOL ]
[ dc CELL ]
[ du UNI ]
[ dm MAT ]
[ dl LAT ]
[ dx XMIN XMAX NX ]
[ dy YMIN YMAX NY ]
[ dz ZMIN ZMAX NZ ]
[ dn TYPE MIN1 MAX1 N1 MIN2 MAX2 N2 MIN3 MAX3 N3 ] 1/2 [ dn TYPE N1 N2 N3 LIM11...LIM1N+1 LIM21...LIM2N+1 LIM31...LIM3N+1 ] 3/4
[ dh TYPE X0 Y0 PITCH N1 N2 ZMIN ZMAX NZ ]
[ dumsh UNI NC CELL0 BIN0 CELL1 BIN1 ... ]
[ de EGRID ]
[ di TBIN ]
[ ds SURF DIR ]
[ dir COSX COSY COSZ ]
[ dtl SURF ]
[ df FILE FRACTION ]
[ dt TYPE PARAM ]
[ dhis OPT ]
[ dfl FLAG OPT ]
[ da MAT FLX ]
[ dfet TYPE PARAMS ]
[ dphb PHB ]
[ dmesh MESH ]
Detector definition. The first parameter:
| PART | : particle type (n = neutron, p = photon) |
is optional in single particle simulations. The remaining parameters are defined by separate key words followed by the input values.
Detector response (dr):
| MT | : response number |
| MAT | : material name or "void" if the material at the collision point is used |
Notes:
- If the detector is assigned with multiple responses, the results are divided correspondingly into separate bins.
- The response numbers are ENDF reaction MT's and special reaction types.
- Positive response numbers are associated with microscopic cross sections and the result is independent of material density. Materials for microscopic cross sections must consist of a single nuclide.
- Microscopic reactions to ground and isomeric states can be calculated by adding "g" or "m" at the end of the reaction number (e.g. 102g and 102m refer to radiative capture to ground and isomeric states, respectively). This option is available only for nuclides with branching ratios.
- Negative response numbers are associated with macroscopic cross sections and special types, and the result is multiplied by material density.
- The response material in the dr entry must not be confused with the material in the dm entry. The former defines the material for the response function, while the latter determines the volume of integration.
- By default, Serpent allows a detector to have at most 10,000,000 bins.
Detector volume (dv):
| VOL | : volume in cm3 (3D geometry) or cross-sectional area in cm2 (2D geometry) |
Notes:
- The results are divided by detector volume, which is by default set to 1.
Detector cell (dc):
| CELL | : cell name where the detector is scored |
Notes:
- If multiple detector cells are defined, the results are correspondingly divided into multiple bins.
Detector universe (du):
| UNI | : universe name where the detector is scored |
Notes:
- If multiple detector universes are defined, the results are correspondingly divided into multiple bins.
Detector material (dm):
| MAT | : material name where the detector is scored |
Notes:
- If multiple detector materials are defined, the results are correspondingly divided into multiple bins.
- The material entry defines the volume of integration, which must not be confused with the response material in the dr entry.
Detector lattice (dl):
| LAT | : lattice name where the detector is scored |
Notes:
- The lattice detector automatically divides the results into multiple bins corresponding to the lattice cells.
Cartesian mesh detector (dx, dy and dz):
| XMIN | : minimum x-coordinate of the detector mesh |
| XMAX | : maximum x-coordinate of the detector mesh |
| NX | : number of x-bins |
| YMIN | : minimum y-coordinate of the detector mesh |
| YMAX | : maximum y-coordinate of the detector mesh |
| NY | : number of y-bins |
| ZMIN | : minimum z-coordinate of the detector mesh |
| ZMAX | : maximum z-coordinate of the detector mesh |
| NZ | : number of z-bins |
Notes:
- The mesh detectors can be used to sub-divide the results into multiple spatial bins. For a Cartesian mesh the division is provided with separate entries in x-, y- and z- locations.
Curvilinear and unevenly-spaced mesh detector (dn):
| TYPE | : Type of curvilinear mesh - 1 = cylindrical (dimensions r, θ, z), 2 = spherical (dimensions r, θ, φ), 3 = unevenly-spaced orthogonal (dimensions x, y, z), 4 = unevenly-spaced cylindrical (dimensions r, θ, z) |
| MINn | : Minimum value of coordinate n for the mesh division (lengths in cm, angles in degrees). |
| MAXn | : Maximum value of coordinate n for the mesh division (lengths in cm, angles in degrees). |
| Nn | : Number of bins in the n coordinate direction (the radial division will be equal r, not equal volume, in evenly-spaced types 1/2). |
| LIMnm | : Mesh boundary m in the n coordinate direction (lengths in cm, angles in degrees). |
Notes:
- All parameters must be provided, even for one- or two-dimensional curvilinear meshes.
- The results are not divided by cell volume (difference to MCNP mesh tally).
- By default, the curvilinear mesh detectors use the global (universe 0) coordinate system for scoring. If the TYPE parameter is given as a negative value (e.g. -1) the lowest level coordinates are used instead.
- The syntax for curvilinear (evenly-spaced) mesh detectors (1/-1, 2/-2) differs from the unevenly-spaced mesh ones (3/-3, 4/-4).
Hexagonal mesh detector (dh):
| TYPE | : Type of hexagonal mesh (2 = flat face perpendicular to x-axis, 3 = flat face perpendicular to y-axis) |
| X0, Y0 | : coordinates of mesh center |
| PITCH | : mesh pitch |
| N1, N1 | : mesh size |
| ZMIN | : minimum z-coordinate of the detector mesh |
| ZMAX | : maximum z-coordinate of the detector mesh |
| NZ | : number of z-bins |
Notes:
- All parameters must be provided, even for a two-dimensional hexagonal meshes.
Unstructured mesh detector (dumsh):
| UNI | : universe of the unstructured mesh based geometry |
| NC | : number of mesh cell bins included in the output |
| CELLn, BINn | : cell-bin index pairs defining the mapping |
Notes:
- The polyhedral cells in unstructured mesh based geometries are indexed.
- This detector option allows collecting results from the cells into an arbitrary number of bins. One or multiple cells can be mapped into a single bin.
Detector energy binning (de):
| EGRID | : energy grid name |
Notes:
- The results are divided into multiple energy bins based on the grid structure.
- Energy grid structures are defined using the ene card. Pre-defined energy group structures can not be directly used in detectors, they have to be redefined using for example the fourth type of ene card.
- The energy boundaries of photon photon pulse-height and photon heat analog detectors are solely defined by the associated energy grid and not limited by the unionized energy grid defining the model. That means that analog detectors might collect scores below the physics model minimum energy bound, without a cut-off, if the energy grid sets it.
Detector time binning (di):
| TBIN | : time bin structure name |
Notes:
- The results are divided into multiple time bins.
- Time bin structures are defined using the tme card.
- Time bin division may require adjusting the average collision distance (set cfe option) to achieve sufficient statistical accuracy.
Surface current / flux detector (ds):
| SURF | : surface name |
| DIR | : direction with respect to surface normal (-2 = flux, -1 = inward current, 1 = outward current, 0 = net current) |
Notes:
- With this option the detector calculates the particle flux over or current through a given surface.
- The surface flux mode is invoked by setting the direction parameter to -2, otherwise this parameter defines the current direction with respect to surface normal.
- Responses are not allowed with current detectors, and with flux detectors, the material name at the collision point has to be specified ("void" is not allowed).
- The use of single-bin mesh and cell detectors is allowed to define the integration surface of the detector, from version 2.1.32 on.
- The surface is treated separate from the geometry, and its position is always relative to the origin of the root universe. This is the case even if the surface is part of the geometry in another universe.
- The results are integrated over the surface area (other detectors integrate over volume).
Detector direction vector (dir):
| COSY | : component of the direction vector parallel to x-axis |
| COSY | : component of the direction vector parallel to y-axis |
| COSZ | : component of the direction vector parallel to z-axis |
Notes:
- This option multiplies the detector scores with the scalar product between the particle direction of motion and the given direction vector.
Super-imposed track-length detector (dtl):
| SURF | : surface inside which the detector is scored |
Notes:
- This option can be used to apply the track-length estimator for calculating reaction rates inside regions defined by a single surface (sphere, cylinder, cuboid, etc.)
- The purpose of the track-length detector is to provide better statistics for special applications (activation wire measurements, etc.).
- The surface is treated separate from the geometry, and its position is always relative to the origin of the root universe. This is the case even if the surface is part of the geometry in another universe.
Detector file (df):
| FILE | : file name where the scored points are written |
| FRAC | : fraction of recorded scores and ascii/binary option (positive value = ascii, negative value = binary) |
Notes:
- This option can be used to write the scored points in a file.
- When used with the surface current detector this option can provide surface source distributions for other calculations.
- The fraction parameters gives the probability that the score is written in the file and it can be used to reduce the file size in long simulations.
- Source files can be read using the sf entry of source cards.
Special types (dt):
| TYPE | : special type (see below) |
| PARAM | : additional parameters |
The types are:
| -1 | = cumulative spectrum |
| -2 | = division by energy width |
| -3 | = division by lethargy width |
| -4 | = sum over cell or material bins |
| -5 | = importance weighting |
| -6 | = sum over number of scores |
| 2 | = multiply result with another detector defined by PARAM |
| 3 | = divide result with another detector defined by PARAM |
| 4 | = multiply response function by (local) temperature |
Notes:
- Types -1, -2 and -3 are used with energy binning.
- Type -4 can be used to calculate sum over multiple cell or material bins defined using the dc and dm options. By default separate bins are used for each entry.
- Type 3 can be used to calculate flux-weighted averages (microscopic and macroscopic cross sections, etc.).
- When the results are multiplied or divided by another detector, the number of bins must be compatible (single value or matching number of bins).
History collection option (dhis):
| OPT | : option to collect histories (0 = no, 1 = yes) |
Notes:
- When this option is set, the batch-wise results are printed in the history output file, [input]_stats.m.
- Note to developers: statistical tests should be documented
Detector flagging (dfl):
| FLAG | : flag number (between 1 and 64) |
| OPT | : flagging option (0 = reset if scored, 1 = set if scored, -2/2 score if set -3/3 score if not set) |
Notes:
- Detector flagging allows limiting detector scores to histories which have already contributed to another score.
- The first two options reset or set the flag if the detector is scored, respectively. The remaining options test if the flag is set and score the detector accordingly. Positive values apply OR-type logic (detector is scored if any of the associated flags is set/unset) and negative values AND-type logic (detector is scored if all the associated flags are set/unset).
Activation detector (da):
| MAT | : activated material |
| FLX | : flux applied to activation |
Notes:
- Activation detector allows performing activation calculation for materials that are not part of the geometry. The flux spectrum applied to neutron irradiation is taken from the detector scores. The absolute flux level can be set using the FLX parameter. If this parameter is set to -1, also the flux magnitude is taken from the detector scores.
- Requires neutron transport simulation and burnup mode. The material provided with the entry must be burnable, and cannot part of the actual geometry. Volume of the material must be defined using the dv parameter.
- Since the activated material is not part of the physical geometry, this option should be applied only to small samples and other activation calculations in which the isotopic changes do not significantly affect the neutronics.
Functional Expansion Tally detector (dfet):
| TYPE | : functional expansion type |
| PARAMS | : other options, specific to each functional expansion type TYPE |
| Geometry | PARAMS | TYPE | Description | Functional Series | Indexing |
|---|---|---|---|---|---|
| Cartesian | XMIN XMAX XORDER YMIN YMAX YORDER ZMIN ZMAX ZORDER | 1 | Legendre only | 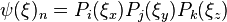
|
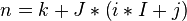
|
| Cylindrical | RMAX RORDER HMIN HMAX HORDER HORIENTATION | 2 | .. | .. | .. |
Notes:
- "-1" can be supplied as an ORDER PARAM to use the built-in default values
- It is not recommended to configure a single FET detector to span multiple different material regions—use individual detectors for each region instead
- Specifics of this implementation:
- The FETs are based on nonseparable expansions, i.e. fully-convolved cross terms are included
- For example, the Legendre-based Cartesian FET uses with
 as a linear indexer of
as a linear indexer of 
- Due to the properties of orthogonality, these cross terms can be neglected in post-analyses if only separable terms are desired
- For example, the Legendre-based Cartesian FET uses
- A generalization of the Euler formulas for any orthogonal functional series is used
- The generated FET coefficients
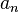 already have all contributions from the orthonormalization constant pre-included, i.e.
already have all contributions from the orthonormalization constant pre-included, i.e. 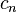 from
from 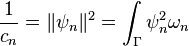
- Thus, an FET can be simply reconstructed/sampled from the standard functional series as:
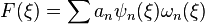
- The generated FET coefficients
- The FETs are based on nonseparable expansions, i.e. fully-convolved cross terms are included
- From version 2.2.0 and on, FET-based detectors follow the standard normalization set in the calculation. The volume standards for detectors are set as default value for FET-based detectors, meaning detectors are not divided by the physical volume (allowing the use of volume detector dv).
- In version 2.2.0, the relative error evaluation associated with FET-based detectors has been revisited.
Detector pulse-height energy broadening (dphb):
| PHB | : user-defined (Gaussian) energy broadening for pulse-height detector function name |
Notes:
- User-defined Gaussian energy broadening functions for pulse height detector are defined using the phb card.
Detector spatial integration domain and binning based on a generic data mesh (dmesh):
| MESH | : name of the datamesh to use for defining the spatial integration domain and binning for the detector scores |
Notes:
- Output mesh index will be flattened (one dimensional).
div (divisor definition)
div MAT [ sep LVL ]
[ subx NX XMIN XMAX ] equal volume [ subr -NX X1 R2 ... XN+1 ] manually spaced limits
[ suby NY YMIN YMAX ] equal volume [ suby -NY Y1 Y2 ... YN+1 ] manually spaced limits
[ subz NZ ZMIN ZMAX ] equal volume [ subz -NZ Z1 Z2 ... ZN+1 ] manually spaced limits
[ subr NR RMIN RMAX ] equal volume [ subr -NR R1 R2 ... RN+1 ] manually spaced limits
[ subs NS S0 ] equal volume [ subs -NS S1 S2 ... SN+1 ] manually spaced limits
[ lims FLAG ]
Divides a material into a number of sub-zones. Input values:
| MAT | : name of the divided material |
| LVL | : geometry level at which the cell-wise division takes place (0 = no division, 1 = last level, 2 = 2nd last level, etc.) |
| NX | : number of x-zones |
| XMIN | : minimum x-coordinate (cm) |
| XMAX | : maximum x-coordinate (cm) |
| Xn | : x-coordinate boundaries (cm) |
| NY | : number of y-zones |
| YMIN | : minimum y-coordinate (cm) |
| YMAX | : maximum y-coordinate (cm) |
| Yn | : y-coordinate boundaries (cm) |
| NZ | : number of z-zones |
| ZMIN | : minimum z-coordinate (cm) |
| ZMAX | : maximum z-coordinate (cm) |
| Zn | : z-coordinate boundaries (cm) |
| NR | : number of radial zones |
| RMIN | : minimum radial coordinate (cm) |
| RMAX | : maximum radial coordinate (cm) |
| Rn | : radial coordinate boundaries (cm) |
| NS | : number of angular sectors |
| S0 | : zero position of angular division (degrees) |
| Sn | : angular-sector boundaries (degrees) |
| FLAG | : flag for mapping regions outside (material) limits to divide material - on (1/yes) or off (0/no) |
Notes:
- The automated divisor feature can be used to sub-divide burnable materials into depletion zones, but the use is not limited to burnup mode.
- The spatial sub-division is based on either Cartesian or cylindrical mesh.
- Volumes of the divided materials must be set manually (see detailed description on the definition of material volumes).
- Using automated instead of manual depletion zone division saves memory, which may become significant in very large burnup calculation problems (see detailed description on memory usage).
- For more information see detailed description on automated depletion zone division.
- The usage of LVL is explained on page automated depletion zone division.
- The feature of mapping regions outside limits is set by default OFF.
- The manually-spaced angular-sector boundaries Sn should cover the full/360 degrees angular space.
- If a material is not divided, all occurrences of it are treated as a single depletion zone. For example, if there are multiple fuel pins with same fuel material type, and no div card is present, all pins are depleted as a single pin.
dtrans (detector mesh transformation)
See transformations.
ene (energy grid definition)
ene NAME 1 E0 E1 ...
ene NAME 2 N Emin Emax
ene NAME 3 N Emin Emax
ene NAME 4 GRID
Defines an energy grid structure. Input values:
| NAME | : energy grid name |
| Ei | : bin boundaries (type 1) |
| N | : number of equi-width bins (types 2 and 3) |
| Emin | : minimum energy (types 2 and 3) |
| Emax | : maximum energy (types 2 and 3) |
| GRID | : name of the pre-defined grid (type 4) |
Notes:
- The first input parameter gives the type (1 = arbitrarily defined, 2 = equal energy-width bins, 3 = equal lethargy-width bins, 4 = pre-defined energy group structure).
- Energy grid structures are used for several purposes, most commonly with detectors.
- See separate description of pre-defined energy group structures.
ftrans (fill transformation)
See transformations.
fun (function definition)
fun NAME TYPE [ ... ]
Defines a function that can be used with detector responses. Input values:
| NAME | : function name |
| TYPE | : function type (currently only supported type is 1 = point-wise tabular data) |
The syntax for type 1 is:
fun NAME 1 INTT X1 F1 X2 F2 ...
where:
| INTT | : is the interpolation type (1 = histogram, 2 = lin-lin, 3 = lin-log, 4 = log-lin, 5 = log-log) |
| Xi, Fi | : are the tabulated variable-value pairs |
Notes:
- The defined function is linked to detector response using response number -100 (syntax: dr -100 NAME).
- The defined function currently is only supported as a flux-based function, aka, flux multiplier.
hisv (history variation matrix definition)
hisv [ BU1 NBR1 BR1,1 BR1,2 ... BR1,NBR1 ]
[ BU2 NBR2 BR2,1 BR2,2 ... BR2,NBR2 ]
...
Defines the history variation matrix for the automated burnup sequence. Input values:
| BUn | : burnup steps at which the branches are invoked |
| NBRn | : number branches in the n-th burnup step |
| BRn,i | : name of the i-th branch in the n-th burnup step |
Notes:
- The automated burnup sequence defined by the hisv card follows the same principle as the coef input card.
- The hisv card performs multiple depletions within a single depletion calculation following the historical variation sequence, performing a restart at each of the listed burnup points, where it applies the variations defined in the listed branches for the given burnup point.
- Positive values in the burnup vector are interpreted as (MWd/kgU), negative values are interpreted as time steps in days.
- The hisv card is used together with the branch card.
ifc (interface file)
ifc FILE [setinmat NMAT MAT1 MAT2 ... MATNMAT ]
[setoutmat NMAT MAT1 MAT2 ... MATNMAT ]
Links a multi-physics interface file to be used with the current input. Input values:
| FILE | : path to the multi-physics interface file |
The optional cards are explained below.
setinmat adds the possibility to link multiple input materials to the same interface, i.e. the same interface gives temperatures and densities (density factors) for multiple materials.
setoutmat adds the possibility to link multiple output materials to the same interface, i.e. the same interface gives temperatures and densities (density factors) for multiple materials.
| NMAT | : number of materials to link to the interface |
| MATi | : name of the ith material linked to the interface |
Notes:
- If multiple materials are linked to the interface using the setinmat/setoutmat option, the densities in the interface file must be given as density factors, i.e. relative to the material card density (values between 0 and 1).
- If the interface is not updated, setinmat/setoutmat options are not eligible. In the case of regular mesh-based, additionally to not updating the interface: a) the input materials cannot be specified using setinmat if power is tallied in pin-type objects; b) the output materials cannot be specified using setoutmat if power is not tallied on the same mesh.
- Option setinmat was setmat in versions before 2.1.32.
- See also Coupled multi-physics calculations.
include (read another input file)
include FILE
Reads another input file. Input values:
| FILE | : name of the input file |
Notes:
- The include card can be used to simplify the structure of complicated inputs.
- The input parser starts reading and processing the new file from the point where the input card is placed. Processing of the original file continues after the new file is completed.
- The included file must contain complete input cards and options, it cannot be used to read the values of another card.
lat (regular lattice definition)
See also Section 3.6 of Serpent 1 User Manual.
lat UNI TYPE X0 Y0 NX NY PITCH UNI1 UNI2 ...
Defines a finite two-dimensional lattice in xy-plane with square or X- or Y-type hexagonal elements. The lattice is infinite in z-direction. Input values:
| UNI | : universe name of the lattice |
| TYPE | : lattice type |
| X0 | : x-coordinate of the lattice origin (origin is in the center of the lattice). |
| Y0 | : y-coordinate of the lattice origin (origin is in the center of the lattice). |
| NX | : number of lattice elements in x-direction |
| NY | : number of lattice elements in y-direction |
| PITCH | : lattice pitch |
| UNIn | : list of universes filling the lattice positions |
Possible lattice types are:
| Type | Description |
|---|---|
| 1 | Square lattice |
| 2 | X-type hexagonal lattice |
| 3 | Y-type hexagonal lattice |
| 14 | X-type triangular lattice |

Notes:
- Number of universes in list of universes must be NX × NY.
- For square lattices the x coordinate increases from left to right and the y coordinate increases from top to bottom, so the first NX values in the list of universes create the bottommost (minimum y) row from minimum x to maximum x and the last NX values in the list of universes create the topmost (maximum y) values. Example of the indexing is provided in the attached figure.
- The line breaks usually present in the list of universes are only used to help visualizing the universe order for the user. Serpent ignores them when processing the list of universes.
- The input of X- and Y-type hexagonal lattices is similar to each other, only the directions of the x- and y-axis change. The axis directions can be checked by using the geometry plotter. Examples of the indexing are provided in the attached figures.
lat UNI TYPE X0 Y0 PITCH UNI1
Defines an infinite two-dimensional lattice in xy-plane with infinitely repeating square or X- or Y-type hexagonal element. The lattice is infinite in z-direction.
| UNI | : universe name of the lattice |
| TYPE | : lattice type |
| X0 | : x-coordinate of the lattice origin |
| Y0 | : y-coordinate of the lattice origin |
| PITCH | : lattice pitch |
| UNI1 | : universe name of the universe filling all lattice positions |
Possible lattice types are:
| Type | Description |
|---|---|
| 6 | Square lattice |
| 7 | Y-type hexagonal lattice |
| 8 | X-type hexagonal lattice |
Notes:
- The order of X- and Y-type hexagonal lattice type numbers is reversed when compared with finite hexagonal lattices.
lat UNI TYPE X0 Y0 NR NS,1 RADIUS1 THETA1 UNI1,1 UNI2,1 ... NS,2 RADIUS2 THETA2 UNI1,2 UNI2,2 ... ...
Defines a finite two-dimensional circular cluster array lattice in xy-plane. The lattice is infinite in z-direction.
| UNI | : universe name of the lattice |
| TYPE | : lattice type |
| X0 | : x-coordinate of the lattice origin |
| Y0 | : y-coordinate of the lattice origin |
| NR | : number of rings in the array |
| NS,R | : number of sectors in Rth ring |
| RADIUSR | : central radius of Rth ring |
| THETAR | : angle of rotation of Rth ring in degrees |
| UNIN,R | : list of universes filling the sector positions in Rth ring |
Possible lattice type is:
| Type | Description |
|---|---|
| 4 | Circular cluster array |
Notes:
- The circular cluster array can be used to define fuel assemblies used for example in AGR, CANDU, MAGNOX and RBMK reactors. It can also be used to define fuel rod layout used for example in TRIGA reactors.
lat UNI TYPE X0 Y0 NL Z1 UNI1 Z2 UNI2 ...
Defines a finite one-dimensional vertical stack in z-direction. The stack is infinite in xy-plane.
| UNI | : universe name of the lattice |
| TYPE | : lattice type |
| X0 | : x-coordinate of the lattice origin |
| Y0 | : y-coordinate of the lattice origin |
| NL | : number of lattice elements in z-direction (number of axial layers) |
| Zn | : z-coordinate of the nth lattice element (lower boundary of the axial layer) |
| UNIn | : universe name filling the nth lattice position |
Possible lattice type is:
| Type | Description |
|---|---|
| 9 | Vertical stack |
Notes:
- The z-coordinates must be given in ascending order.
- Space below the lowest z-coordinate is not defined.
- The top layer fills the entire space above the highest z-coordinate.
- The number of Zn-UNIn pairs must be NL.
lat UNI TYPE X0 Y0 Z0 NX NY NZ PITCHX PITCHY PITCHZ UNI1 UNI2 ...
Defines a finite three-dimensional lattice in xyz-space with cuboidal or X- or Y-type hexagonal prism elements.
| UNI | : universe name of the lattice |
| TYPE | : lattice type |
| X0 | : x-coordinate of the lattice origin |
| Y0 | : y-coordinate of the lattice origin |
| Z0 | : z-coordinate of the lattice origin |
| NX | : number of lattice elements in x-direction |
| NY | : number of lattice elements in y-direction |
| NZ | : number of lattice elements in z-direction |
| PITCHX | : lattice pitch in x-direction |
| PITCHY | : lattice pitch in y-direction |
| PITCHZ | : lattice pitch in z-direction |
| UNIn | : list of universes filling the lattice positions |
Possible lattice types are:
| Type | Description |
|---|---|
| 11 | Cuboidal lattice |
| 12 | X-type hexagonal prism lattice |
| 13 | Y-type hexagonal prism lattice |
Notes:
- Number of universes in list of universes must be NX × NY × NZ.
- For hexagonal prism lattices the x- and y-direction pitches must be equal.
- The universe indexing is the similar as with lattice types 1-3. The lowermost z-level is given first, and the uppermost z-level is given last.
ltrans (lattice transformation)
See transformations.
mat (material definition)
See Chapter 4 of Serpent 1 User Manual.
mat NAME DENS [ tmp TEMP ] [ tms TEMP ] [ tft TMIN TMAX ] [ rgb R G B ] [ vol VOL ] [ mass MASS ] [ burn NR ] [ fix ID TEMP ] [ moder THNAME ZA ] NUC1 FRAC1 [ NUC2 FRAC2 ] [ ... ]
Mandatory information:
| NAME | : Name of the material |
| DENS | : Density of the material (positive for atomic, negative for mass density) or sum to calculate the density from given nuclide fractions |
| NUCn | : Identifier of nth nuclide in composition, e.g. "92235.03c" or "U-235.03c". |
| FRACn | : Fraction of nth nuclide in composition, positive values are interpreted as atomic fractions/densities, negative values as mass fractions/densities. |
Optional cards:
tmp: Material temperature for Doppler-preprocessor
| TEMP | : Temperature (in Kelvin) of the material for Doppler-broadening preprocessor |
tms: Material temperature for on-the-fly TMS temperature treatment
| TEMP | : Temperature (in Kelvin) of the material for on-the-fly TMS temperature treatment |
tft: Temperature limits for material for coupled multi-physics calculations
| TMIN | : Lower limit for material temperature |
| TMAX | : Upper limit for material temperature |
rgb: Material color for geometry plots
| R | : Value for the red channel of geometry plots (between 0 and 255) |
| G | : Value for the green channel of geometry plots (between 0 and 255) |
| B | : Value for the blue channel of geometry plots (between 0 and 255) |
vol: Material volume
| VOL | : Volume of the material in cm3 (3D geometry) or cross-sectional area in cm2 (2D geometry) |
mass: Material mass
| MASS | : Mass of the material in grams |
burn: Flag material for depletion
| NR | : Set to 1 in order to deplete material. The depletion zone division should be done using the div-card. |
fix: Library information for decay nuclides
| LIB | : Library ID (e.g. "09c") for nuclides without cross section data. |
| TEMP | : Temperature (in Kelvin) for nuclides without cross section data. |
moder: Use thermal scattering data library for a nuclide. The moder entry can be used several times to define thermal scattering libraries for multiple nuclides, such as hydrogen and deuterium in heavy water.
| THNAME | : Name of the thermal scattering data library defined using the therm card. |
| ZA | : Nuclide ZA of the thermal scatterer (e.g. 1001 for H-1). |
Notes:
- This description is incomplete for both the descriptions and optional settings.
- See defining material volumes and set mvol regarding other ways to set the material volumes for example in burnup calculations.
- The nuclide identifier for nuclides with associated cross-sections corresponds to ZZAAA.ID and, for nuclides without associated cross-sections, e.g., decay nuclides, to ZZAAAI. The identifiers include Z, the atomic number; A, the mass number of the nuclide; I, the isomeric state (0 = ground state, 1 = metastable state); and ID, the library identifier. For nuclides without associated cross-sections, include the fix option to indicate the library and temperature of the given nuclides.
mesh (mesh plot definition)
mesh ORI XPIX YPIX [ SYM MIN1 MAX1 MIN2 MAX2 MIN3 MAX3 ]
mesh 8 CMAP DET ORI XPIX YPIX [ SYM MIN1 MAX1 MIN2 MAX2 MIN3 MAX3 ]
mesh 10 ORI XPIX YPIX
Produces a png-format mesh plot of various results. Input values:
| ORI | : orientation with respect to coordinate axes |
| XPIX | : horizontal image size in pixels |
| YPIX | : vertical image size in pixels |
| SYM | : symmetry option (not used in Serpent 2) |
| MINn MAXn | : boundaries of the plotted region |
| CMAP | : color map used for plotting detector scores (positive entry for linear, negative for log-scale) |
| DET | : detector name |
Notes:
- The first format produces a mesh plot where fission rate and thermal flux distribution are plotted using hot and cold color schemes, respectively. This type of mesh plot is convenient for illustrating the neutronics of thermal systems. The orientation parameter defines the coordinate axis perpendicular to the plot plane: 1 - x-axis (projection on yz-plane); 2 - y-axis (projection on xz-plane); 3 - z-axis (projection on xy-plane).
- The second format can be used for plotting scores that contribute to a detector. The additional input parameters are the detector name and the color map used in the plot.
- The third format generates a mesh-plot of the temperature distribution. This can be a good way to check the temperature distribution, provided by an external solver, during a coupled calculation.
- The color maps are: 1 - hot; 2 - cold; 4 - jet; 5 - black and white; 6 - hsv; 7 - spring; 8 - summer; 9 - autumn; 10 - winter; 11 - green-purple; 12 - purple-orange; 13 - blue-red. Many of these correspond to what is used in Matlab. Logarithmic scale is used if the number is given with a minus sign.
- Detector scores are collected in the mesh cells (see the detector card and the list of ENDF reaction MT's and special reaction types for more information). The distribution is scaled according to the minimum and maximum values.
- Some special detector types, such as pulse-height detectors and analog photon heating detectors cannot be associated with mesh plots.
- The mesh plot always produces results that are integrated over space. If no boundaries are provided, the integration is carried over the entire geometry.
- Setting the orientation parameter of a detector mesh plot to 4 produces a plot in cylindrical coordinates. Instead of Cartesian boundaries the entered values are then the radius, angle and axial coordinate.
- The symmetry option was used in Serpent 1. The parameter must be provided for Serpent 2 as well, even though it is not used. The value can be set to zero.
- Mesh plot produced by the nth mesh-card is written in file [input]_mesh[n].png.
mflow (material flow definition)
mflow NAME NUC1 λ1 [ NUC2 λ2 ] [ ... ]
Defines the material flow. Input values:
| NAME | : name of the material flow |
| NUCn | : identifier of nth nuclide in composition |
| λn | : reprocessing constant of nth nuclide in composition (in s-1) |
Notes:
- The nuclide ID can be replaced with "all", in which case all nuclides are included with the same reprocessing fraction λ.
- The nuclide ID should follow the ZAI or ISO format (e.g., 922350 or U-235).
mix (mixture definition)
mix NAME [ rgb R G B ]
[ vol VOL ]
[ mass MASS ]
MAT1 F1
MAT2 F2
...
Defines a mixture of two or several materials. Input values:
Mandatory information:
| MATn | : material name |
| Fn | : material fraction (positive values for volume, negative values for mass fractions) |
Optional cards:
rgb: Material color for geometry plots
| R | : Value for the red channel of geometry plots (between 0 and 255) |
| G | : Value for the green channel of geometry plots (between 0 and 255) |
| B | : Value for the blue channel of geometry plots (between 0 and 255) |
vol: Material volume
| VOL | : Volume of the material in cm3 (3D geometry) or cross-sectional area in cm2 (2D geometry) |
mass: Material mass
| MASS | : Mass of the material in g |
Notes:
- Mixtures can be used to define complicated material definitions consisting of two or more physical materials mixed homogeneously.
- Serpent decomposes these mixtures into standard materials before running the transport simulation.
- The decomposed material compositions can be written into file using the -mix command line option.
- Nuclide specific thermal scattering data is automatically brought from component materials to the mixture.
- Many other input cards such as set trc, set iter nuc, sens pert matlist are not automatically inherited by the mixture from the components and should be directly defined using the mixture material name (opposed to component material names) if they are to be applied to the mixture.
nest (nested universe definition)
nest U TYPE [ MAT1 R1 ] [ MAT2 R2 ] ... [ MATN ]
nest U [ MAT1 TYPE1 PARAM11 PARAM12 ... ] [ MAT2 TYPE2 PARAM21 PARAM22 ... ] ... [ MATN ]
Defines a universe consisting of nested regions. Input values:
| U | : universe name |
| TYPE | : nested surface type (single surface for all regions) |
| MAT1 ... MATN | : material regions |
| R1 ... RN-1 | : outer radii |
| TYPE1 ... TYPEN-1 | : nested surface type (different surfaces for each region) |
| PARAMnm ... | : surface parameters |
Notes:
- The nest card defines an entire universe consisting of nested material regions. The boundaries are defined by surfaces nested inside each other. The outermost region is infinite.
- The material entries can be replaced by fill U0, in which case the region is filled by another universe.
- The first format allows defining nests in which all surfaces are of same type and centred at the origin. Only surfaces that are characterized by a single outer radius are accepted (cylinders, spheres and some regular prisms). The pin and particle definitions are short-hand notations of the nest card.
- The second format allows mixing different surface types. In this case all surface parameters need to be provided after the surface type.
particle (particle geometry definition)
particle U
[ MAT1 R1 ]
[ MAT2 R2 ]
...
[ MATN ]
Defines a particle universe. Input values:
| U | : universe name |
| MAT1 ... MATN | : material regions |
| R1 ... RN-1 | : outer radii |
Notes:
- The particle card defines an entire universe consisting of nested spherical shells. The boundaries are defined by sphere surfaces. The outermost region is radially infinite.
- The material entries can be replaced by fill U0, in which case the region is filled by another universe.
- Most typically used for defining TRISO fuel particles.
- The particle card is special case of a nested universe type.
- See also description of explicit stochastic geometry type.
pbed (explicit stochastic (pebble bed) geometry)
pbed U0 Ubg FILE [ OPT ]
Defines a stochastic particle / pebble-bed geometry. Input values:
| U0 | : universe name for the dispersed medium |
| Ubg | : background universe, i.e. universe filling the space between particles / pebbles |
| FILE | : input file containing the particle/pebble data |
| OPT | : additional options (currently only pow to produce power distribution in a separate output file) |
The syntax of the file containing the particle/pebble data is:
X1 Y1 Z1 R1 U1
X2 Y2 Z2 R2 U2
...
Where:
| Xn, Yn, Zn | : are the coordinates |
| Rn | : is the radius |
| Un | : is the universe |
Notes:
- Creates a universe (U0), which is filled with spherical sub-universes for which the coordinates are read from a separate file.
- The coordinates can be defined manually, or using the automated disperser routine.
- Can be used for modelling stochastic particle / pebble-bed geometries in multiple levels.
- If the power distribution option is set, the pebble/particle-wise distribution is written in file [U0].out.
- See also HTGR geometry examples.
phb (pulse-height Gaussian energy broadening definition)
phb NAME TYPE [ ... ]
Defines a user-defined (Gaussian) energy broadening function for pulse-height detector (dphb). Input values:
| NAME | : pulse-height (Gaussian) energy broadening function name |
| TYPE | : pulse-height function type (1 = energy-resolution interpolation, 2 = energy-FWHM interpolation, 3 = energy-resolution fitting, 4 = energy-FWHM fitting) |
The syntax for the different types is as follows:
phb NAME 1 INTT Emax,1 R1 Emax,2 R2 ...
where:
| INTT | : is the interpolation type (currently only supported type is 2 = lin-lin interpolation data) |
| Emax,i, Ri | : are the maximum energy-resolution tabulated pairs |
Notes:
- Full width at half maximum is calculated as:

- Energies should be given in ascending order.
phb NAME 2 INTT Emax,1 FWHM1 Emax,2 FWHM2 ...
where:
| INTT | : is the interpolation type (currently only supported type is 2 = lin-lin interpolation data) |
| Emax,i, FWHMi | : are the maximum energy-full width at half maximum pairs |
Notes:
- Energies should be given in ascending order.
phb NAME 3 a b
where:
| a, b | : are the parameters to define the energy resolution fit: 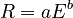
|
phb NAME 4 a b c
where:
| a, b, c | : are the parameters to define the energy full width at half maximum fit: 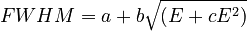
|
pin (pin geometry definition)
pin U [ MAT1 R1 ] [ MAT2 R2 ] ... [ MATN ]
Defines a pin universe. Input values:
| U | : universe name |
| MAT1 ... MATN | : material regions |
| R1 ... RN-1 | : outer radii |
Notes:
- The pin card defines an entire universe consisting of nested annular material regions. The boundaries are defined by axially infinite cylindrical surfaces. The outermost region is radially infinite.
- The material entries can be replaced by fill U0, in which case the region is filled by another universe.
- Most typically used for defining fuel pins, but can also be applied to guide tubes, control rods, etc.
- The pin card is special case of a nested universe type.
plot (geometry plot definition)
plot TYPE XPIX YPIX [ POS MIN1 MAX1 MIN2 MAX2 ]
plot TYPE Fmin Fmax E XPIX YPIX [ POS MIN1 MAX1 MIN2 MAX2 ]
Produces a png-format geometry plot. Input values:
| TYPE | : defines the plot type (orientation and plotting of boundaries) |
| XPIX | : horizontal image size in pixels |
| YPIX | : vertical image size in pixels |
| POS | : position of plot plane |
| MIN1 | : minimum horizontal coordinate of plotted region |
| MAX1 | : maximum horizontal coordinate of plotted region |
| MIN2 | : minimum vertical coordinate of plotted region |
| MAX2 | : maximum vertical coordinate of plotted region |
| Fmin | : minimum importance for importance map plots |
| Fmax | : maximum importance for importance map plots |
| E | : particle energy for importance map plots |
Notes:
- The TYPE parameter consists of one or two concatenated values ('AB'):
- The first value ('A') defines the plot plane (1 = yz, 2 = xz, 3 = xy).
- The second value ('B') defines which boundaries are plotted (0 = no boundaries, 1 = cell boundaries, 2 = material boundaries, 3 = both). If the second value in is not provided, material boundaries are plotted.
- Importance maps read using the wwin card can be plotted on top of the geometry by setting the second value ('B') of the type parameter to 4 (linear color scheme) or 5 (logarithmic color scheme) for cell importances, and to 6 (linear color scheme) or 7 (logarithmic color scheme) for source importances. The input parameters then also include the minimum and maximum importance and the particle energy. If importance maps are provided for both neutrons and photons, they can be plotted by entering positive and negative energy values, respectively.
- If both, minimum and maximum importance values are set to "-1", Serpent automatically adjusts them based on the weight-window mesh data, from version 2.2.0 and on.
- Each material plotted with different color. The colors are sampled randomly, unless defined using the rgb entry in the material card.
- Void is plotted in black and special colors are used to plot geometry errors (red = overlap, green = undefined region).
- The position parameter defines the location of the plot plane on the axis perpendicular to it (e.g. z-coordinate for xy-type plot).
- The minimum and maximum coordinates define the boundaries of the plotted region (e.g. minimum and maximum x- and y-coordinates for xy-type plot). If these coordinates are not provided, the plot is extended to the maximum dimensions of the geometry.
- The relative dimensions of image size in pixels should match that of the plotted region. Otherwise the image gets distorted.
- Geometry plotter requires compiling the source code with GD Graphics libraries.
- Command line parameter -plot stops the execution after the geometry plots are produced. Option -qp invokes a quick plot mode, which does not check for overlaps. Option -noplot skips the geometry plots altogether.
- See also detailed description on geometry plotter.
- Geometry plot produced by the nth plot-card is written in file [input]_geom[n].png.
- Note to developers: particle type should be included as an input parameter in importance map plots.
rep (reprocessor definition)
rep NAME [ rc SRC TGT MFLOW MODE ] [ rm MAT1 MAT2 ] [ ru UNI1 UNI2 ]
Defines the reprocessing controllers. Input values:
| NAME | : name of the reprocessor. |
| SRC | : name of the source material, from which the flow is moved |
| TGT | : name of the target material, to which the flow is moved |
| MFLOW | : name of the material flow |
| MODE | : continuous reprocessing mode |
| MAT1 | : name of the replaced material |
| MAT2 | : name of the replacing material |
| UNI1 | : name of the replaced universe |
| UNI2 | : name of the replacing universe |
Notes:
- The reprocessor name identifies the reprocessing regime in the depletion calculation dep card. The syntax corresponds to dep pro NAME.
- The nuclides identifier of those included in both source SRC and target TGT materials in reprocessors should follow the same format, either ZA.ID or ISO.ID (for nuclides with cross sections), or ZAI (for nuclides without associated cross sections, and adding the fix entry to the mat card).
- The (continuous) reprocessing implementation works with materials, not universes. Therefore, define the universes associated with those burnable materials as surface-cell type universes.
- Multiple reprocessing controllers/regimes can be defined within the same reprocessor definition.
- The rc continuous reprocessing option can be used to define the material flow between the source and the target materials.
- The material flow is defined using the mflow card.
- The continuous reprocessing MODE defines how to incorporate the material flow into the Bateman equations:
MODE Material source Material target Material flow 0 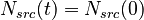
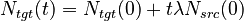
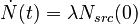
1 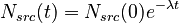
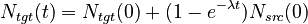
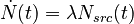
2 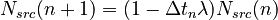
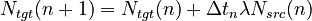
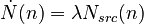
- MODE 0 : no changes at the source material and adds λN0 from the source material to the target material when solving the Bateman equations (N0 are initial compositions).
- MODE 1 : subtracts λN from the source material and adds it to the target material when solving the Bateman equations.
- MODE 2 : subtracts λNn from the source material and adds it to the target material when solving the Bateman equations (compositions updated with each burnup step, n).
- The rm material reprocessing option replaces one material with another, MAT1 by MAT2.
- The ru universe reprocessing option replaces one universe with another, UNI1 by UNI2.
sample (Temperature / density data sample definition)
sample NX XMIN XMAX NY YMIN YMAX NZ ZMIN ZMAX
Samples values from the initial material temperatures and densities to a file using a Cartesian grid.
Input values:
| NX | : Number of values to sample in the x-direction. |
| XMIN | : Minimum coordinate to sample from in the x-direction. |
| XMAX | : Maximum coordinate to sample from in the x-direction. |
| NY | : Number of values to sample in the y-direction. |
| YMIN | : Minimum coordinate to sample from in the y-direction. |
| YMAX | : Maximum coordinate to sample from in the y-direction. |
| NZ | : Number of values to sample in the z-direction. |
| ZMIN | : Minimum coordinate to sample from in the z-direction. |
| ZMAX | : Maximum coordinate to sample from in the z-direction. |
Notes:
- The data from each sample is written in a separate [input]_sampleN.m file.
- Positive values for the density data correspond to atomic densities, while negative values correspond to mass densities.
- Materials with no temperature specified either in their mat-card or through an interface definition will show a temperature of 0.
sens (sensitivity calculation definition)
sens pert
sens resp
sens opt
Definitions for the perturbations, responses and options for sensitivity calculations.
solid (irregular 3D geometry definition)
solid 1 UNI BGUNI MESH_SPLIT MESH_DIM SZ1 SZ2 ... SZMESH_DIM POINTS_FILE FACES_FILE OWNER_FILE NEIGHBOUR_FILE MATERIALS_FILE
Creates an unstructured mesh-based geometry universe. Input values are:
| UNI | : universe name for the irregular geometry |
| BGUNI | : name of the background universe filling all undefined space |
| MESH_SPLIT | : Splitting criterion for the adaptive search mesh (maximum number of geometry cells in search mesh cell) |
| MESH_DIM | : number of levels in the adaptive search mesh |
| SZi | : Size of the search mesh at level i |
| POINTS_FILE | : Path to the unstructured mesh points file |
| FACES_FILE | : Path to the unstructured mesh faces file |
| OWNER_FILE | : Path to the unstructured mesh owner file |
| NEIGHBOUR_FILE | : Path to the unstructured mesh neighbour file |
| MATERIALS_FILE | : Path to the unstructured mesh materials file |
solid 2 UNI BGUNI MESH_SPLIT MESH_DIM SZ1 SZ2 ... SZMESH_DIM MODE R0 body BODY1 CELL1 MAT1 file BODY1 FILE1 SCALE1 X1 Y1 Z1 file BODY1 FILE2 SCALE2 X2 Y2 Z2 ... body BODY2 CELL2 MAT2 file BODY2 FILE3 SCALE3 X3 Y3 Z3 file BODY2 FILE4 SCALE4 X4 Y4 Z4 ...
Creates an STL-based geometry universe. Input values are:
| UNI | : universe name for the irregular geometry |
| BGUNI | : name of the background universe filling all undefined space |
| MESH_SPLIT | : Splitting criterion for the adaptive search mesh (maximum number of geometry cells in search mesh cell) |
| MESH_DIM | : number of levels in the adaptive search mesh |
| SZi | : Size of the search mesh at level i |
| MODE | : Mode for handling the triangulated geometry (1 = "fast", 2 = "safe"). |
| R0 | : Radius inside which two points of the STL-geometry are joined into one. |
| BODYi | : Name of solid body i |
| CELLi | : Name of geometry cell i linked with body i |
| MATi | : Material filling cell i |
| FILEi | : Path to a file containing an STL solid model, multiple files can be linked to one body |
| SCALEi | : Scaling factor for the STL model in FILEi |
| Xi | : Shift in x-direction to the STL model in FILEi |
| Yi | : Shift in y-direction to the STL model in FILEi |
| Zi | : Shift in z-direction to the STL model in FILEi |
Notes:
- The material entries can be replaced by fill UNIi, in which case the cell i is filled by the given universe.
solid 3 INTERFACE_FILE
Creates an unstructured mesh-based geometry universe with unstructured mesh-based temperature and/or density distributions. Input values are:
| INTERFACE_FILE | : Path to the interface file containing the rest of the parameters |
Notes:
- For simple example of a CAD-based geometry, see the Stanford critical bunny.
- For more information on the unstructured mesh based geometry see Unstructured mesh based input.
- For a simple example of an unstructured mesh based geometry see the Simple umsh 8 cubes input.
src (source definition)
src NAME [ PART ]
[ sw WGT ]
[ sc CELL ]
[ su UNI ]
[ sm MAT ]
[ sp X Y Z ]
[ sx XMIN XMAX ]
[ sy YMIN YMAX ]
[ sz ZMIN ZMAX ]
[ srad RMIN RMAX ]
[ ss SURF ]
[ sd U V W ]
[ se E ]
[ sb N INTT E1 WGT1 E2 WGT2 ... ]
[ sr NUC MT ]
[ st TMIN TMIN ]
[ sf FILE TYPE ]
[ si N P1 P2 ... ]
[ sg MAT MODE ]
Source definition. The first parameter:
| PART | : particle type (n = neutron, p = photon) |
is optional in single particle simulations. The remaining parameters are defined by separate key words followed by the input values.
Source weight (sw):
| WGT | : relative source weight |
Notes:
- When multiple sources are defined, each definition is sampled with equal probability. This probability can be changed by assigning different weights for each source.
- The weights are automatically normalized before the calculation is started.
Source cell (sc):
| CELL | : cell inside which the source points are sampled |
Notes:
- Setting a source cell is one of the options that can be applied to define the spatial distribution of source particles.
- The selection is based on rejection sampling, and if the source cell occupies a small volume of the geometry, the sampling efficiency can be increased by defining a bounding box/(vertical) cylinder around the cell (using the sx, sy and sz or sp, srad and sz options, respectively).
- If no spatial distribution is defined, particles are sampled uniformly over the geometry.
Source universe (su):
| UNI | : universe inside which the source points are sampled |
Source material (sm):
| MAT | : material inside which the source points are sampled |
Notes:
- Setting a source material is one of the options that can be applied to define the spatial distribution of source particles.
- The selection is based on rejection sampling, and if the source material occupies a small volume of the geometry, the sampling efficiency can be increased by defining a bounding box/(vertical) cylinder around the cell (using the sx, sy and sz or sp, srad and sz options, respectively).
- If no spatial distribution is defined, particles are sampled uniformly over the geometry.
Source point (sp):
| X, Y, Z, | : coordinates of the source point |
Notes:
- Setting a point source is one of the options that can be applied to define the spatial distribution of source particles.
- If no spatial distribution is defined, particles are sampled uniformly over the geometry.
Source boundaries (sx, sy, sz and srad):
| XMIN, XMAX | : Boundaries on X-axis |
| YMIN, YMAX | : Boundaries on Y-axis |
| ZMIN, ZMAX | : Boundaries on Z-axis |
| RMIN, RMAX | : Radial boundaries |
Notes:
- Source boundaries are used to define a bounding box/(vertical) cylinder inside which the source particles are sampled.
- The radial boundaries are centered around the point defined by sp and can be used in combination with sz.
- Can be used in combination with cell and material sources to increase the sampling efficiency.
- If no bounding box is defined, particles are sampled uniformly over the geometry.
Surface source (ss):
| SURF | : surface on which the source particles are sampled |
Notes:
- The surface source is currently limited to infinite vertical cylinder (cyl) and sphere (sph) surface types.
- Particles are started in the direction of the inward surface normal.
Source direction (sd):
| U, V, W, | : direction vector of source particles |
Notes:
- The source direction option can be set to define a unidirectional source.
- If no directional dependence is defined, the direction of source particles is sampled isotropically.
Source energy (se):
| E | : energy of source particles |
Notes:
- The source energy option can be used to define a monoenergetic source.
- The default energy of neutrons and photons is 1 MeV.
- This option can also be used together with the source reaction option (sr).
Source energy bins (sb):
| N | : number of bins |
| INTT | : interpolation (0 = line spectrum, 1 = histogram, 2 = lin-lin, 4 = log-lin) |
| En | : upper boundary of the energy bin |
| WGTn | : weight of the energy bin |
Notes:
- This option allows defining an arbitrary source spectrum in the form of tabular data.
- The bins are entered in the order of ascending energy, and weight of the first bin must be set to zero.
- Interpolation is given in a separate parameter from version 2.1.31 on.
- Here, a simple test input that demonstrates the source spectrum definition.
Source reaction (sr):
| NUC | : nuclide name |
| MT | : reaction number |
Notes:
- The source reaction determines a distribution function for source energy (for example, 235U fission spectrum can be defined as: sr 92235.09c 18).
- The reaction numbers are ENDF reaction MT's, and the data is obtained from standard cross section libraries.
- Applies to neutrons only.
- When the source energy parameter (se) is defined, the value is used as the energy of the incoming neutrons.
Source time (st):
| TMIN, TMAX | : time boundaries |
Notes:
- This parameter defines a time interval for the sampled source particles. The starting time is sampled uniformly between the given minimum and maximum.
- All source particles are started at time zero by default.
Source file (sf):
| FILE | : file path to source file |
| TYPE | : file type (-1 = binary, 1 = ASCII) |
Notes:
- Source files allow defining arbitrary distributions by reading the particle coordinates, direction, energy, weight and time from a file.
- Source files can be produced using the df entry of detector cards, or the set csw or set gsw options.
User-defined source routine (si):
| N | : number of parameters |
| Pn | : parameters passed as arguments into the subroutine |
Notes:
- This option allows defining an arbitrary source distributions with a user-defined subroutine.
- The source parameters are passed as arguments into the subroutine, together, with sampled position, direction energy, weight and time.
- For complete description see source file "usersrc.c".
- The subroutine may be overwritten with the blank template file when installing updates.
Radioactive decay source (sg):
| MAT | : material name or -1 |
| MODE | : sampling mode (1 = analog, 2 = implicit) |
Notes:
- Radioactive decay source combines material compositions to decay data read from ENDF format libraries and forms the normalized source distribution automatically.
- Material compositions can be defined manually, or read from binary restart files produced by a burnup or activation calculation (see the set rfw and set rfr options).
- If the material name is replaced by -1, source points are started from all radioactive materials.
- The analog sampling mode preserves the average number of particles produced in radioactive decay, but may lead to poor sampling efficiency in geometries with both low and high-active materials.
- The implicit sampling mode preserves the total statistical weight of emitted particles and produces a uniform source distribution over activated materials.
- In version 2.1.28 the source is limited to photon line spectra.
- The calculation produces an additional output file [input]_gsrc.m or [input]_nsrc.m that contains the gamma/neutron source spectra, respectively.
- See practical example for more information.
srtrans (source transformation)
See transformations.
strans (surface transformation)
See transformations.
surf (surface definition)
surf NAME TYPE [ PARAM1 PARAM2 ... ]
Defines a surface. Input values:
| NAME | : is the surface name |
| TYPE | : is the surface type |
| PARAMn | : are the surface parameters |
Notes:
- The name is used to identify the surface, for example, in the cell card.
- See separate description on surface types.
- Surfaces can be moved and rotated using transformations.
therm and thermstoch (thermal scattering)
therm NAME LIB
therm NAME TEMP LIB1 LIB2
therm NAME 0 LIB1 LIB2 LIB3 ...
thermstoch NAME TEMP LIB1 LIB2
Defines thermal scattering data that can be linked to nuclides using input entry moder in the material cards. When using thermal scattering data together with TMS on-the-fly temperature treatment, the third input value of the therm card is 0. In this case, Serpent interpolates the thermal scattering data automatically to the local temperature, as defined either using the tms input entry in the material definition (mat card) or via the multi-physics interface (ifc card).
Input values:
| NAME | : name of the thermal scattering data |
| LIBi | : thermal scattering data identifiers as defined in the directory file (acelib) |
| TEMP | : temperature to which the thermal scattering data is interpolated |
Notes:
- When using on-the-fly interpolation of thermal scattering data, LIBi must cover the whole temperature range in which the materials appear in the geometry. I.e. extrapolation of the data is not supported.
- Thermal scattering data is interpolated using the methodology of makxsf code
- The interpolation can be performed using the stochastic mixing approach with thermstoch. This interpolation mode is not available for on-the-fly interpolation.
- The continuous S(α, β) formalism is available from version 2.1.32 on.
- Version 2.2.0 includes the on-the-fly temperature treatment for the continuous S(α,β) formalism.
tme (time binning definition)
tme NAME 1 LIM1 LIM2 ...
tme NAME 2 NB Tmin Tmax
tme NAME 3 NB Tmin Tmax
Defines a time binning structure. The second entry sets the binning type (1 = arbitrary, 2 = uniform, 3 = log-uniform). Remaining values:
| NAME | : name of the time binning |
| NB | : number of bins |
| LIMn | : time bin boundaries in arbitrary binning |
| Tmin | : minimum time boundary in uniform or log-uniform binning |
| Tmax | : maximum time boundary in uniform or log-uniform binning |
Notes:
- The entered values are in seconds
- The first limit in the arbitrary type (type = 1), is the lower bound of the first bin. The second limit is the upper bound of the first bin and so on.
- Time binning is used with detectors and dynamic simulation mode.
trans (transformations)
trans TYPE UNIT [ IDX ] LVL
trans TYPE UNIT [ IDX ] X Y Z
trans TYPE UNIT [ IDX ] X Y Z θx θy θz ORD
trans TYPE UNIT [ IDX ] X Y Z α1 α2 α3 α4 α5 α6 α7 α8 α9 ORD
trans TYPE UNIT [ IDX ] rot X0 Y0 Z0 I J K β
Defines surface, universe, fill, lattice, detector mesh or source transformation. Input values:
| TYPE | : type of transformation (S = surface, F = fill, U = universe, L = lattice, D = detector mesh, SR = source) |
| UNIT | : surface, cell, universe, lattice, detector mesh or source name to which the transformation is applied |
| IDX | : index number in lattice transformation (type L) |
| LVL | : level number in universe level transformation |
| X,Y,Z | : translation vector |
| θx θy θz | : rotation angles with respect to x-, y- and z-axes (in degrees) |
| α1 ... α9 | : coefficients of the rotation matrix |
| ORD | : order in which translations and rotations are applied (1 = rotations first, 2 = translations first) |
| X0,Y0,Z0 | : Origin of vector defining rotation axis. |
| I,J,K | : Components of vector defining rotation axis. |
| β | : Angle around rotation axis defined by a vector (in degrees). NB: In Serpent 2.1.29 positive values correspond to rotation to the negative mathematical direction and vice versa. |
Notes:
- Fill transformation is applied in the universe filling the given cell.
- Level transformation is a special type of universe transformation, in which the coordinates in the given universe are obtained relative to geometry level LVL.
- Lattice transformation requires to provide the index for the transformation IDX.
- Source transformation is inverted. transformation is inverted compared to how surface, universe, etc. are handled.
- By default translations are applied before rotations, and the order can be switched using the ORD parameter.
- Rotations can be defined either by providing the three angles with respect to the three coordinate axes, or by defining the rotation matrix. In the second case Serpent applies vector multiplication
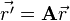 where
where 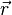 and
and 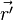 are the position vectors before and after the operation and coefficients α1 ... α9 define the 3 by 3 matrix
are the position vectors before and after the operation and coefficients α1 ... α9 define the 3 by 3 matrix 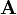 .
. - To preserve backwards compatibility, input parameters "strans", "utrans", "ftrans", "ltrans", "dtrans" and "srtrans" without the following type identifier are also accepted for defining surface, universe, fill, lattice, detector mesh and source transformations, respectively. To preserve compatibility with Serpent 1, parameter "trans" without type identifier defines a universe transformation.
transb (burnup transformation)
transb STEP [ <trans> ]
Defines burnup-dependent surface, universe or fill transformation. Input values:
| STEP | : step in units of burnup (positive values) or days (negative values) |
| <trans> | : list of parameters associated with the transformation |
Notes:
- The parameters associated with the transformation follow the standard transformation cards syntax without "trans" identifier.
transv and transa (velocity and acceleration transformations)
transv TYPE UNIT [ IDX ] [ tlim T0 T1 TTYPE ] VX VY VZ
transa TYPE UNIT [ IDX ] [ tlim T0 T1 TTYPE ] AX AY AZ
Defines surface, universe, fill, lattice, detector mesh or source transformation. Input values:
| TYPE | : type of transformation (S = surface, F = fill, U = universe, L = lattice, D = detector mesh, SR = source) |
| UNIT | : surface, cell, universe, lattice, detector mesh or source name to which the transformation is applied |
| IDX | : index number in lattice transformation (type L) |
| T0 | : beginning time of the transformation |
| T1 | : end time of the transformation |
| TTYPE | : transformation type after end time (1 = movement stops, 2 = transformation removed, 3 = initial acceleration and velocity removed, but velocity accumulated due to acceleration remains) |
| VX,VY,VZ | : Initial velocity vector |
| AX,AY,AZ | : Initial acceleration vector |
Notes:
- Fill transformation is applied in the universe filling the given cell.
- The transformation is updated at the simulation time-interval boundaries.
- See UGM 2016 Moving geometry.
- See Rotating Translating STL Bunny.
umsh (unstructured mesh-based geometry definition)
UNI BGUNI MESH_SPLIT MESH_DIM SZ1 SZ2 ... SZMESH_DIM POINTS_FILE FACES_FILE OWNER_FILE NEIGHBOUR_FILE MATERIALS_FILE
Defines an unstructured mesh-based geometry. Input values:
| UNI | : universe name for the unstructured mesh-based geometry |
| BGUNI | : name of the background universe filling all undefined space |
| MESH_SPLIT | : splitting criterion for the adaptive search mesh (maximum number of geometry cells in search mesh cell) |
| MESH_DIM | : number of levels in the adaptive search mesh |
| SZi | : size of the search mesh at level i |
| POINTS_FILE | : path to the unstructured mesh points file |
| FACES_FILE | : path to the unstructured mesh faces file |
| OWNER_FILE | : path to the unstructured mesh owner file |
| NEIGHBOUR_FILE | : path to the unstructured mesh neighbour file |
| MATERIALS_FILE | : path to the unstructured mesh materials file |
Notes:
- See also description of solid (irregular 3D geometry definition), type 1.
utrans (universe transformation)
See transformations.
wwgen (response matrix based importance map solver)
wwgen NAME LIM NI MOD ERG MSH
MIN1 MAX1 SZ1
MIN2 MAX2 SZ2
MIN3 MAX3 SZ3
DET1 W1 [ DET2 W2 ... ]
wwgen NAME LIM NI MOD ERG MSH
SZ1 SZ2 SZ3
LIM11 LIM12 ...
LIM21 LIM22 ...
LIM31 LIM32 ...
DET1 W1 [ DET2 W2 ... ]
wwgen NAME LIM NI MOD ERG MSH
X0 Y0
P NX NY
MIN3 MAX3 SZ3
DET1 W1 [ DET2 W2 ... ]
Defines the parameters for importance map calculation. Input values:
| NAME | : a unique name to identify the calculation |
| LIM | : convergence criterion (typical value 1E-12) |
| NI | : maximum number of iterations |
| MOD | : solution mode (1 = single detector, 2 = multiple detectors, 3 = global variance reduction) |
| ERG | : energy group structure (or -1 if no energy dependence is included) |
| MSH | : mesh type (1 = Cartesian, 2 = Cylindrical, 4 = x-type hexagonal, 5 = y-type hexagonal, 6 = unevenly-spaced xyz, 8 = unevenly spaced cylindrical) |
| MINn | : minimum mesh boundary (nth coordinate) |
| MAXn | : maximum mesh boundary (nth coordinate) |
| SZn | : number of mesh cells (nth coordinate) |
| LIMnm | : mesh boundary mth (nth coordinate) |
| X0, Y0 | : mesh center of hexagonal mesh (currently must be centered at the origin) |
| P | : hexagonal cell pitch |
| NX, NY | : hexagonal mesh size |
| DETi | : detectors used as target response functions |
| Wi | : weight factors for detector scores |
Notes:
- The solution mode provides various options on how the responses are used for calculating the importances.
- The detector entries can be left out in global variance reduction mode (MOD = 3), in which case the mesh is optimized to uniformly populate the entire geometry.
- Cartesian and cylindrical mesh are defined by outer mesh boundaries and number of mesh cells.
- Unevenly-spaced meshes are defined by providing the mesh cell boundaries separately.
- The coordinate axes 1, 2 and 3 in Cartesian mesh refer to (x,y,z) and in cylindrical mesh to (r,θ,z), with θ given in degrees.
- The hexagonal mesh is defined by mesh center, cell pitch, number of cells in the radial dimensions (similar to the hexagonal lattice) and axial binning.
- The mesh must be defined slightly larger than the geometry (the mesh boundaries should not coincide with the geometry boundaries).
- Source points located on mesh cell boundaries cause fatal errors.
- May not work if source distribution is biased with weight.
- The importance mesh is printed in file [input].wwd.
- Importance (weight window) meshes are read using the wwin card.
- See also practical examples on Variance reduction.
- This capability is still very much under development. The input syntax may be revised at some point.
wwin (weight window mesh definition)
wwin NAME [ wf FILE FMT ] [ wn F X Y Z E ] [ wx C G ] [ wt SB TYPE MIN MAX ] [ wi ITP NI WWG1 DF1 WWG2 DF2 ... ] [ wi ITP NI WWG NX NY NZ NLOOP NTRK ISPL NSPL DSPL1 SX1 SY1 SZ1 DSPL2 SX2 SY2 SZ2 ...]
Defines a weight window mesh for variance reduction. The first parameter:
| NAME | : a unique name to identify the mesh |
identifies the mesh. The remaining parameters are defined by separate key words followed by the input values.
Notes:
- Only works in external source simulation mode.
- Importance (weight window) meshes can be generated by running the response matrix based solver, or read in MCNP WWINP format.
- Importance maps can be visualized using the geometry plotter.
- See also set wwb and set maxsplit for setting options for weight windows, splitting and Russian roulette.
- See also practical examples on Variance reduction.
- This capability is still very much under development. The input syntax may be revised at some point.
Mesh file (wf):
| FILE | : file path and name of the importance mesh file |
| FMT | : file format (1 = mesh produced by Serpent importance map generator, 2 = MCNP WWINP format weight window mesh file) |
Notes:
- By default the importance map is read from the mesh file and used as-is, the additional options are provided for adjustments.
- Currently the MCNP format only supports simple mesh types (no sub-mesh).
Mesh normalization (wn):
| F | : importance for renormalization |
| X,Y,Z | : coordinates of point used for renormalization |
| E | : energy used for renormalization |
Notes:
- The importances can be renormalized by fixing the value at a given position and energy.
Mesh adjustment (wx):
| C | : constant multiplier for adjusting importances |
| G | : exponential for adjusting importances |
Notes:
- The importances can be adjusted by constant multiplier C and exponential factor G such that
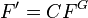 .
.
Types and options (wt):
| SB | : option to set source biasing on (1/yes) or off (0/no) with Serpent-generated importance maps |
| TYPE | : bounds type for Serpent-generated weight-windows (1 = averaged, 2 = segment-wise) |
| MIN | : minimum truncation limit for importances |
| MAX | : maximum truncation limit for importances |
Notes:
- Source biasing is currently not available
Weight-window iterations, fixed mesh (wi):
| ITP | : iteration type (1 = fixed mesh) |
| NI | : number of iterations between Monte Carlo simulation and the response matrix solver |
| WWGi | : name of the WWG-structure used in the iteration |
| DFi | : global density factor |
Notes:
- The fixed mesh option (ITP = 1) allows performing iterations using a single or multiple meshes generated using the response matrix based solver.
- The global density factor is a multiplier applied to all material densities.
Weight-window iterations, adaptive mesh (wi):
| ITP | : iteration type (2 = geometry-based adaptation, 3 = tracking-based adaptation) |
| NI | : number of iterations between Monte Carlo simulation and the response matrix solver |
| WWG | : name of the WWG-structure used in the iteration |
| NX | : number of x-divisions for the adaptive mesh |
| NY | : number of y-divisions for the adaptive mesh |
| NZ | : number of z-divisions for the adaptive mesh |
| NLOOP | : number of outer iteration loops in generation of adaptive mesh |
| NTRK | : number of tracks per loop in generation of adaptive mesh |
| ISPL | : importance split criterion |
| NSPL | : neighbor split criterion |
| DSPLi | : density split criterion (negative values for mass, positive values for atomic density) |
| SZi | : minimum cell dimension |
Notes:
- The adaptive mesh option (ITP = 2 or 3) starts with a coarse base mesh, and refines the resolution iteratively.
- There are two adaptive mesh options. In the geometry-based option (ITP = 2) Serpent covers the geometry with NTRK random tracks and splits cells according to density criteria. In the tracking-based option (ITP = 3) the tracks are started from the source instead. The procedure is repeated NLOOP times.
- Cell splitting is defined using the NX, NY and NZ options. For example NX = 2, NY = 2, NZ = 2 results in each cell being split to 8 sub-cells (octree mesh). For 2D meshes the NZ parameter must be set to 1.
- Splitting is carried out recursively, until limiting criteria are met. The DSPL parameters define upper density boundaries and minimum cell sizes for stopping the splits.
- The importance split criterion defines the maximum relative difference between the importances of two adjacent cells. If the criterion is not met, both cells are split.
- The neighbor split criterion defines the maximum number of neighbor allowed for a cell. If the criterion is not met, the cell is split.
Input options
Input options are used to set various calculation parameters that are not included in the main input cards. Each option is identified by key word "set". Optional values are enclosed within square brackets.
set absrate
set absrate A [ MAT ]
Sets normalization to total absorption rate.
| F | : number of neutrons absorbed per second (neutrons/s) |
| MAT | : dummy parameter |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- Absorption includes all reactions in which the incident neutron is lost, i.e. all capture reactions and fission.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set powdens, set flux, set genrate, set fissrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set acelib
set acelib LIB1 [ LIB2 LIB3 ... ]
Sets the cross section directory file paths. Input values:
| LIBn | : file paths to directory files |
Notes:
- If the file path contains special characters it is advised to enclose it within quotes.
- A default directory path can be set by defining environment variable SERPENT_DATA. The code looks for cross section directory files in this path if not found at the absolute.
- A default cross section directory file can be set by defining environment variable SERPENT_ACELIB. This file will be used if no other path is given with set acelib.
set adf
set adf UNI SURF SYM [ENF]
Sets parameters for the calculation of assembly discontinuity factors (ADFs). Input values:
| UNI | : universe where spatial homogenization is performed |
| SURF | : surface enclosing the universe |
| SYM | : symmetry option (see separate list) |
| ENF | : option to skip diffusion flux solver and enforce flat homogeneous flux distribution based on mean heterogeneous flux (1/yes). Default is (0/no). |
Notes:
- The surface enclosing the universe can be super-imposed (i.e. not part of the geometry definition), but it must enclose the entire universe.
- The surface is super-imposed on the geometry, i.e. its parameters (coordinates) are relative to the root universe (default 0)
- When the universe is surrounded by zero net-current (reflective) boundary conditions, the ADFs are calculated as the ratios of surface- and volume-averaged heterogeneous flux.
- When the net current is non-zero, the calculation is based on the ratio of surface-averaged homogeneous and heterogeneous flux. The homogeneous flux is obtained from a built-in diffusion flux solver.
- Calculation parameters for the diffusion flux solver can be set using the set dfsol option.
- Calculation of ADFs is currently allowed only for planes and infinite square and hexagonal prisms.
- Symmetry options are used to average out the statistical variation in the ADFs, which might otherwise lead to systematic errors in core calculations. It is important that the options are used only when the geometry has the corresponding symmetry.
- See separate description of output parameters in the [input]_res.m.
- The ADFs can be printed to the group constant output with set coefpara.
- ADFs are calculated in the few-group structure used for group constant generation.
- The ENF parameter should be switched on only in rare cases (and you should know what you are doing).
set alb
set alb UNI SURF DIR
Sets parameters for calculating albedos. Input values:
| UNI | : universe where spatial homogenization is performed |
| SURF | : surface for which the albedos are calculated |
| DIR | : current direction (-1 = inward, 1 = outward) |
Notes:
- When this option is set, Serpent calculates both total albedos (ratio of currents) and partial albedos (response matrix).
- The surface enclosing the universe can be super-imposed (i.e. not part of the geometry definition), but it must enclose the entire universe.
- The surface is super-imposed on the geometry, i.e. its parameters (coordinates) are relative to the root universe (default 0)
- The current direction is given relative to the surface normal vectors
- The universe is needed only for labelling the results in the output files.
- See separate description of output parameters.
- Albedos are calculated in the few-group structure used for group constant generation.
set arr
set arr MODEN [ MODEG ]
Sets analog reaction rate calculation on or off. Input values:
| MODEN | : mode for neutrons (0 = no reactions included, 1 = include only reactions that affect neutron balance, 2 = include all reactions) |
| MODEG | : mode for photons (0 = no reactions included, 1 = include all reactions) |
Notes:
- Analog reaction rates are calculated by counting sampled events and printed in a separate output file [input]_arr[bu].m, where "bu" is the burnup step.
- See detailed description on the reaction rate output file.
set ba
set ba ZAI1 ZAI2 ...
Defines isotopes handled separately as burnable absorbers. Input values:
| ZAIn | : nuclide identifiers (ZAI) |
Notes:
- Some burnup applications require separate treatment for isotopes that are used as burnable absorbers but also produced in fission. This input parameter can be used to separate the transmutation chains.
- Isotope handled as the burnable absorber is created by duplicating the original and renaming it as ZAIn + 1000.
- For Gd-155, for example, the fission product isotope would be assigned ZAI 641550 and the burnable absorber ZAI 642550.
- The input parameter defines the entire transmutation chain. Listing Gd-isotopes 641540 641550 641560 641570 641580 creates a transmutation path from Gd-154 to Gd-158. Listing only the main absorbers (641550 641570) produces a different result, since the capture products of Gd-155 and Gd-157 are lost.
set bala
set bala OPT
Sets OpenMP load balancing on or off. Input values:
| OPT | : probability to store particles in common queue (0 = off, >0 = on) |
Notes:
- Load balancing may improve OpenMP parallel scalability in calculations with significant branching (most typically related to coupled neutron/photon calculations or variance reduction).
- The option is ON with OPT= 1 with weight-window/variance reduction calculations and dynamic/time-dependent calculation modes. Otherwise, it is set OFF by default. Before version 2.2.0, the default behavior was always OFF.
- When this option is set, the random number sequence is no longer preserved.
set bc
set bc MODE
Sets the boundary conditions for all outer boundaries of the geometry. Input values:
| MODE | : boundary type (1 = vacuum, 2 = reflective, 3 = periodic) |
set bc MODE ALB
Sets the boundary conditions with albedo for all outer boundaries of the geometry. Input values:
| MODE | : boundary type (1 = vacuum, 2 = reflective, 3 = periodic) |
| ALB | : albedo |
set bc MODEX MODEY MODEZ
Sets the boundary conditions separately for x-, y- and z-directions. Input values:
| MODEX | : boundary type in x-direction (1 = vacuum, 2 = reflective, 3 = periodic) |
| MODEY | : boundary type in y-direction (1 = vacuum, 2 = reflective, 3 = periodic) |
| MODEZ | : boundary type in z-direction (1 = vacuum, 2 = reflective, 3 = periodic) |
set bc MODEX MODEY MODEZ ALB
Sets the boundary conditions with albedo separately for x-, y- and z-directions. Input values:
| MODEX | : boundary type in x-direction (1 = vacuum, 2 = reflective, 3 = periodic) |
| MODEY | : boundary type in y-direction (1 = vacuum, 2 = reflective, 3 = periodic) |
| MODEZ | : boundary type in z-direction (1 = vacuum, 2 = reflective, 3 = periodic) |
| ALB | : albedo |
Notes:
- The boundary conditions can be set either for all directions at once (single parameter) or x-, y- and z-directions separately (three parameters). Albedos are provided by adding one more parameter in the list.
- The default boundary condition is vacuum (= 1) in all directions.
- Albedo boundary conditions are invoked by multiplying the particle weight with factor ALB each time a reflective or periodic boundary is hit.
- Repeated boundary conditions (reflective or periodic) are based on universe transformations, which limits outer boundary to surfaces that form regular lattices (square and hexagonal prisms, rectangles, cubes and cuboids).
- Repeated boundary conditions are applied on the first surface of outside cells (see definition of outside cells in the cell card)
- For symmetry purposes Serpent provides the universe symmetry option.
- For more information, see detailed description on boundary conditions.
- The boundary condition type numbers can also be given as strings, with black = 1, reflective = 2 and periodic = 3.
set blockdt
set blockdt MAT1 MAT2 ...
Defines the list of materials where delta-tracking is never used. Input values:
| MATn | : material names |
Notes:
- This option is used to override selection of tracking mode based on the probability threshold (see set dt) in individual materials.
- Use of delta-tracking can be forced in individual materials using set forcedt.
- For more information on tracking modes, see the detailed description on delta- and surface-tracking.
- Note to developers: should have different lists for neutrons and photons?
set bralib
set bralib LIB1 [ LIB2 LIB3 ... ]
Sets isomeric branching data library file paths. Input values:
| LIBn | : library file paths |
Notes:
- Isomeric branching data libraries are standard ENDF format files containing energy-dependent branching ratios. The data is read from ENDF files 9 and 10.
- Serpent uses constant branching ratios by default. The default values can be overridden using the set isobra option. Energy-dependent data read read from ENDF format files overrides the constant ratios.
- If the file path contains special characters it is advised to enclose it within quotes.
- A default directory path can be set by defining environment variable SERPENT_DATA. The code looks for decay data files in this path if not found at the absolute.
- See also: example input
- Example data from the JEFF-3.1 activation file
set branchless
set branchless OPT [ WGT_LOW WGT_HIGH ]
Option that enables the branchless collision method for variance reduction. Input values:
| OPT | : option to switch calculation on (1/yes) or off (0/no). Default is off. |
| WGT_LOW | : weight lower-boundary (default value: 0.2) |
| WGT_HIGH | : weight upper-boundary (default value: 10.0) |
Notes:
- The branchless algorithm suppresses the variability due to the simultaneous propagation of the several branches associated to a fission event
- The branchless method uses analog scattering combined with forced fission so that after each collision, the neutron is either a scattering neutron or a fission neutron. In a non-multiplying method, the branchless method behaves as implicit capture.
set bumode
set bumode MODE [ ORDER SSD ]
Sets the burnup calculation mode. Input values:
| MODE | : burnup calculation mode |
| ORDER | : CRAM order |
| SSD | : number of substeps for CRAM decay steps (default or 0: use TTA) |
The possible settings for mode are:
| Mode | Description |
|---|---|
| 1, tta | Transmutation Trajectory Analysis (TTA) |
| 2, cram | Chebyshev Rational Approximation Method (CRAM) |
The CRAM order parameter can only be given when choosing the CRAM mode. The possible settings for CRAM order are:
| CRAM order |
|---|
| 2 |
| 4 |
| 6 |
| 8 |
| 10 |
| 12 |
| 14 |
| 16 |
| -16 |
| -48 |
Notes:
- The default setting for the burnup calculation mode is CRAM.
- Default value for the CRAM order is 14 resulting in order 14 PFD CRAM.
- Negative values of CRAM order mean using IPF form of CRAM with order of the absolute value of the parameter.
- Positive values refer to PFD form of CRAM.
- Decay calculations (see dep (depletion history)) and burnup calculations with very low flux are always calculated with TTA disregarding this input before version 2.1.32. The latter, very low flux condition, only applies to calculations not involving continuous reprocessing.
- Positive values of SSD enforce usage of CRAM with given number of substeps.
- The Serpent 1 MODE 3, a variation TTA method, in which cyclic transmutation chains are handled by inducing small variations in the coefficients instead of solving the extended TTA equations, is overwritten by the standard TTA method MODE 1.
- Version 2.2.0 includes the sub-step method for depletion calculations involving continuous reprocessing.
set bunorm
set bunorm NORM
Sets the burnup calculation normalization mode if it is not bound to a single material. Input values:
| NORM | : burnup calculation normalization mode (1 = all materials, 2 = burnable materials, 3 = non-burnable materials) |
Notes:
- The default normalization for burnup calculations includes all materials.
set ccmaxiter
set ccmaxiter NITER
Sets the maximum number of coupled calculation iterations. Input values:
| NITER | : number of iterations. |
Notes:
- Default maximum number of iterations is 1 (no iteration).
- The iteration is stopped when either the maximum number of iterations or the maximum active neutron population (set with set ccmaxpop) has been simulated.
- See Coupled multi-physics calculations for further information.
set ccmaxpop
set ccmaxpop CPOP
Sets the maximum total live population to simulate in a coupled calculation. Input values:
| CPOP | : total active population to simulate. |
Notes:
- Default maximum population is INFTY/1e6.
- The iteration is stopped when either the maximum number of iterations (set with set ccmaxiter) or the maximum active neutron population has been simulated.
- Only the population simulated during active cycles is included in this amount.
- This is mostly useful if the neutron population per iteration is not constant.
- See Coupled multi-physics calculations for further information.
set cdop
set cdop OPT
Sets the Doppler broadening method for the energy spectrum of the scattered photons. Input values:
| OPT | : option to set Doppler broadening method off (0/no) or on (1/yes). The default option is on. |
Notes:
- If the Doppler broadening method is switched off, the incoherent scattering function approximation is used for calculating the energy.
- In both cases, the direction of the photon is calculated using the incoherent scattering function.
set cea
set cea OPT
Sets the Compton electron angular distribution model on and off. Input values:
| OPT | : option to set the Compton electron angular distribution model off (0/no) or on (1/yes). The default option is on. |
Notes:
- Electron travels in the direction of the momentum transfer vector. This is equal to the free-electron scattering angle when Doppler broadening is not used.
set cfe
set cfe LN [ TN LG TG ]
Defines the minimum mean distance for scoring the collision flux estimator (CFE) for photons and neutrons. Input values:
| LN | : minimum mean distance for scoring the CFE for neutrons |
| TN | : minimum mean time interval for scoring the CFE for neutrons |
| LG | : minimum mean distance for scoring the CFE for photons |
| TG | : minimum mean time interval for scoring the CFE for photons |
Notes:
- The use of delta-tracking necessitates the use of CFE for scoring the integral reaction rates. The scoring is based on both real and virtual collision to improve the statistics in low density regions (and short time intervals).
- The minimum mean distance is the statistical mean-free-path (mfp) of collisions that contribute to the CFE. Collisions are more frequent if the physical mfp is shorter.
- In time-dependent simulations it may be more convenient to define the minimum mean time between two collisions, to get sufficient statistics for short time bins.
- The default minimum mean scoring distance is 20 cm for both neutrons and photons. Adjusting the distance affects both statistics and running time, but it should be noted that no studies have been performed on what the optimal value should be.
- Only one criterion can be provided for each particle type. If distance is given, time must be set to -1 and vice versa.
- For more information on tracking modes and CFE, see the detailed descriptions on delta- and surface-tracking and result estimators.
- The collision flux estimator in Serpent is described in an article in Annals of Nuclear energy from 2017.[2]
- In version 2.1.27 and earlier the name of this input option was "set minxs".
set cmm
set cmm OPT
Sets calculation of diffusion coefficients using the cumulative migration method (CMM) on or off. Input values:
| OPT | : option to switch CMM calculation on (1/yes) or off (0/no) |
Notes:
- Calculation of diffusion coefficients using CMM might take considerable time. This option allows switching the calculation off if the data is not needed.
- The calculation of CMM diffusion coefficients was revised in version 2.1.31 so that the calculated values may be different than with previous versions.
- CMM diffusion coefficients can be calculated also when using implicit capture reactions beginning from version 2.1.31.
- CMM diffusion coefficients and transport cross sections are reasonable only when they are calculated over entire geometry (homogenized region covers the entire geometry and is surrounded by periodic or reflective boundary conditions).
- This means that e.g. pin cell diffusion coefficients can not be calculated from a 2D fuel assembly calculation.
- One may try to approximate the CMM diffusion coefficients with TRC diffusion coefficients with transport correction for hydrogen for light water reactor applications.
- Using private results array may be recommended when CMM diffusion coefficients are calculated.
set coefpara
set coefpara FMT [ PARAM1 PARAM2 ... ]
Defines the parameters included in the separate group constant output file. Input values:
| FMT | : output format, currently used for including or excluding statistical errors (0 = not included, 1 = included) |
| PARAMn | : list of parameters or detectors included in the file |
Notes:
- The group constant output file [input].coe is produced when the automated burnup sequence is invoked.
- The available parameters are listed under homogenized group constants in the description of the [input]_res.m output file.
- Detectors are identified by the name assigned to them in the detector card.
set combing
set combing MODE
Option that enables the combing approach for precursors population control as an alternative to Russian roulette and splitting in dynamic source simulations. Input values:
| MODE | : combing population-control mode (0 = none, 1 = weight-based, 2 = emission-based) |
Notes:
- The combing method can achieve variance reduction and save computer time by keeping the population size approximately constant over time steps. In super-critical systems, it prevents the population from growing without bound while, in sub-critical systems, it does it from dying. In critical systems, it avoids the divergence of the variance of the population due to fluctuations of fission chains.
set comfile
set comfile INFILE OUTFILE
Defines the communication files used in the file-based coupled calculation communications. Input values:
| INFILE | : Path to inwards communication file (signals to Serpent). |
| OUTFILE | : Path to outwards communication file (signals from Serpent). |
Notes:
- Setting up a communication mode will enable the coupled calculation mode.
- The communication options set comfile, set ppid and set pport are mutually exclusive, aka, multiple signalling modes are not allowed.
- For more information see: External coupling
set confi
set confi OPT
Sets confidentiality flag on or off. Input values:
| OPT | : option to set confidentiality flag on (1/yes) or off (0/no) |
Notes:
- This option can be used to label calculations as confidential. If the option is set, text "(CONFIDENTIAL)" is printed in the run-time output next to the calculation title and the value of variable CONFIDENTIAL_DATA in the [input]_res.m output file is set to 1.
set coverxlib
set coverxlib LIB1 [ LIB2 LIB3 ... ]
Sets COVERX-format multi-group covariance data file paths. Input values:
| LIBn | : file paths to multi-group covariance data files in the COVERX format (ASCII or binary) |
Notes:
- If covariance data is linked when running Sensitivity calculations, Serpent will automatically apply the sandwich rule using the calculated sensitivity vectors and propagate the covariance data to uncertainties of the sensitivity responses.
set covlib
set covlib LIB1 [ LIB2 LIB3 ... ]
Sets plain ASCII multi-group covariance data file paths. Input values:
| LIBn | : file paths to multi-group covariance data files in the plain ASCII format (ASCII or binary) |
Notes:
- If covariance data is linked when running Sensitivity calculations, Serpent will automatically apply the sandwich rule using the calculated sensitivity vectors and propagate the covariance data to uncertainties of the sensitivity responses.
set cpd
set cpd DEPTH [ NZ ZMIN ZMAX ] [ LVL1 LVL2 ]
Sets on the calculation of lattice-wise power distributions to output file [input]_core0.m on. Input values:
| DEPTH | : The number of levels included. 1 is the first lattice calculated from universe 0 usually corresponding to assembly-wise distribution. 2 includes the first two levels usually corresponding to the assembly- and pin-wise distributions. |
| NZ | : Number of equal sized axial bins into which the lattices are divided. |
| ZMIN | : Minimum z-coordinate for the axial division. |
| ZMAX | : Maximum z-coordinate for the axial division. |
| LVL1 | : User-defined first level where to define the lattice-wise power distribution |
| LVL2 | : User-defined second level where to define the lattice-wise power distribution |
Notes:
- The default values for NZ, ZMIN and ZMAX are 1, -INFTY and INFTY, respectively.
set cpop
set cpop NPG NGEN NSKIP [ NSKIP2 ]
Sets parameters for simulated neutron population for corrector neutron transport solutions in burnup calculation. Typically used with the SIE burnup scheme. Input values
| NPG | : number of neutrons per generation |
| NGEN | : number of active generations |
| NSKIP | : number of inactive generations |
| NSKIP2 | : number of inactive generations on further iterations for the same burnup point |
Notes:
- As the SIE burnup scheme executes the corrector step multiple times for each burnup step, combining the results from each iteration, it may be a good idea to run more iterations with less active neutron histories per iteration.
set csw
set csw FILE
Writes source points in criticality source simulation into a file. Input values:
| FILE | : file name where the source points are written |
Notes:
- Only source points from active cycles are included.
set dataout
set dataout TABLE_LIST
Defines the tables included in the nuclear and material data file [input].out.m. Input values:
| TABLE_LIST | : list of tables |
Notes:
- The file lists all nuclides and their reactions as they are read from the nuclear data libraries. The material data includes isotopic compositions and densities, as well as volumes and masses if available. (See the description of the nuclear and material data output).
- The data is divided into two sections: nuclear data (Tables 1-4) and material data (Table 5), and the tables are the following:
- Table 1: Summary of nuclide data
- Table 2: Reaction and decay data
- Table 3: Fission yield data (only in burnup mode)
- Table 4: Lost transmutation paths (only in burnup mode)
- Table 5: Summary of material compositions
- The entry-names to input the tables are: "all"/0 = Tables 1-5, "nuc_summary"/1 = Tables 1, "nuc_readec"/2 = Table 2, "nuc_nfy"/3 = Table 3, "nuc_lostpath"/4 = Table 4, "mat_summary"/5 = Table 5, "allnuc"/8 = Tables 1-4, "allmat"/9 = Table 5.
- The default behavior is to include all the tables/data available. To omit the [input].out file, select "-1".
set dbrc
set dbrc Emin Emax NUC1 [ NUC2 ... ]
Enables the use of doppler-broadening rejection correction (DBRC). Input values:
| Emin | : Minimum energy for DBRC |
| Emax | : Maximum energy for DBRC |
| NUCn | : Nuclide identifiers for which to apply DBRC to, with 0 K cross section data, e.g. "92238.00c" |
Notes:
- This description is not complete.
- Use of DBRC requires 0 K cross section data.
- See also Section 5.6 of Serpent 1 User Manual.
- This input could be given without any nuclides before version 2.1.32. Then DBRC was not used at all.
set dd
set dd MODE [ X0 Y0 α0 ]
Invokes domain decomposition. Input values
| MODE | : decomposition mode (0 = none, 1 = simple, 2 = sector, 3 = sector + center) |
| X0 | x-coordinate of the domain decomposition origin (origin is the centre of the angular division) |
| Y0 | y-coordinate of the domain decomposition origin (origin is the centre of the angular division) |
| α0 | angular position of the domain decomposition origin (origin is the initial position of the angular division) |
Notes:
- Domain decomposition works in MPI mode by separating burnable materials into different parallel tasks.
- Number of domains is given by the number of MPI tasks
- Only burnable materials separated into depletion zones using the "div sep" option are decomposed
- MODE 1 decomposes the geometry based on the automatically assigned depletion zone indexes (not recommended).
- MODE 2 decomposes the zones into sectors and MODE 3 adds a central zone if the number of domains is greater than 4.
- The additional input options, X0, Y0 and α0, are available for MODE 2 and 3 (default values are zero).
- Decomposed materials are plotted in domain-specific colors (unless the rgb entry in the material card is used)
- See practical example for more information.
set declib
set declib LIB1 [ LIB2 LIB3 ... ]
Sets the decay data library file paths. Input values:
| LIBn | : library file paths |
Notes:
- Decay libraries are standard ENDF format files containing decay data.
- If the file path contains special characters it is advised to enclose it within quotes.
- A default directory path can be set by defining environment variable SERPENT_DATA. The code looks for decay data files in this path if not found at the absolute.
- From version 2.2.0 and on, a default decay data library directory file can be set by defining environment variable SERPENT_DECLIB. This file will be used if no other path is given with set declib.
set decomp
set decomp OPT [ ELEM1 ELEM2 ... ]
Decomposes elemental entries in material cards into isotopes. Input values:
| OPT | : option to include (1) or exclude (0) elements from decomposed list |
| ELEMn | : element names |
Notes:
- Elemental entries are identifed from zero A in ZA.
- The decomposition is based on built-in isotope fractions.
- If the list is not provided, all elemental entries are decomposed.
set delnu
set delnu OPT
Sets delayed neutron emission on or off. Input values:
| OPT | : option to switch delayed neutron emission on (1/yes) or off (0/no) |
Notes:
- Delayed neutron emission is on by default in neutron criticality source and off by default in (static/dynamic) external source simulation mode.
- In time-dependent calculations, driven by the set dynsrc option, precursor based delayed neutron emission is included in the calculation: off at fission, but on at delayed nubar in total nubar.
- See separate description of physics options in Serpent for differences to other codes.
set depmtx
set depmtx MODE
Print burnup matrixes to [input]_depmtx_[mat]_[bu]_[ss].m file during burnup calculation, where "bu" is the burnup step and "ss" is the substep.
| MODE | : Set printing on (1/yes) or off (0/no). |
Notes:
- With non-constant predictor, this option will stop the simulation up to version 2.1.31.
- With multiple substeps, only the last one is kept after the simulation up to version 2.1.31.
- The burnup matrix output is named depmtx_[mat][bu].m up to version 2.1.31.
set depout
set depout MODE [STEP]
Controls which burnable material compositions are printed into the [input]_dep.m output file in case of divided materials. Input values:
| MODE | : value indicating, which materials to output to the [input]_dep.m file (1 = only partials, 2 = only parents, 3 = both) |
| STEP | : value indicating the print-out interval of the [input]_dep.m file (0 = final step, 1 = all steps, 2 =none) |
Notes:
- Parent materials refer to materials defined by mat cards, and partials to depletion zones created automatically using the div card.
- Default mode is 2 (only parents) and default print-out interval step is 1 (all steps).
- Print-out interval step option 2, no [input]_dep.m generation, can be combined with post-processing re-depletion: "-rdep" command line option.
- This option when is used with the domain decomposition feature, set dd, in a mode different from 2, generates multiple depletion files which are named adding _dd[mpiid] (domain decomposition identifier) to the standard file name. Each of them contains the partial materials information of the given domain/MPI task.
set deppara
set deppara PARAM_LIST
Defines the variables included in the depletion output file [input]_dep.m. Input values:
| PARAM_LIST | : list of variables |
Notes:
- The default behavior is to include the full-set of variables: atomic density, mass density, activity, decay heat, spontaneous fission rate, photon emission rate, ingestion toxicity, and inhalation toxicity (see the description of the depletion output).
- The entry-names to input the variables are: "atom" = atomic density, "mass" = mass density, "activity" = activity, "dh" = decay heat, "sf" = spontaneous fission rate, "gsrc" = photon emission rate, "ingtox" = ingestion toxicity, "inhtox" = inhalation toxicity.
- Alternatively, the "all" entry would include the full-set of variables, and the "none" entry would exclude the full-set of variables.
set depstepbunorm
set depstepbunorm NORM
Sets the depletion step normalization in burnup calculations based on energy deposition. Input values:
| NORM | depletion step normalization mode based on energy deposition (1 = all materials, 2 = burnable materials) |
Notes
- By default, for energy deposition modes 0/1, the normalization includes only "burnable" materials while for energy deposition modes 2/3, the normalization includes "all" materials (see set edepmode).
set dfsol
set dfsol MODE [ DC NP ]
Options for homogeneous diffusion flux solver. Input values:
| MODE | : boundary conditions for solver (1 = include net currents at boundary surfaces and corners, 2 = include only surface currents) |
| DC | : type of diffusion coefficient used in the calculation (1 = INF_DIFFCOEF, 2 = TRC_DIFFCOEF) |
| NP | : number of points for trapezoidal integration for homogeneous flux |
Notes:
- This input option is used to control how the deterministic diffusion flux solver used to obtain assembly discontinuity factors (set adf) and pin power distributions (set ppw) is run.
- Default mode is 1 (include both surfaces and corners in solution).
- Default diffusion coefficient is INF_DIFFCOEF, option 2 requires the set trc option.
- Default number of points for trapezoidal integration is 100.
- See also separate description of the built-in diffusion flux solver.
- The format was revised in update 2.1.27 (DC option was added between MODE and NP).
set dix
set dix OPT
Sets double indexing for cross section energy grid look-up on or off:
| MODE | : option to set double indexing on (1/yes) or off (0/no) |
Notes:
- Double indexing[3] is a method to speed-up the cross section look-up when energy grid unionization is not used for microscopic data.
- The method can be used only in optimization modes 1 and 3 (modes 2 and 4 are based on energy grid unionization).
set dspec
set dspec EGRIDp EGRIDn
Sets the energy grid structure for decay spectra.
| EGRIDp | : energy grid structure for photons |
| EGRIDn | : energy grid structure for neutrons |
Notes:
- The photon/neutron decay spectra is printed in the [input]_gsrc.m or [input]_nsrc.m output file, respectively.
- The energy group spectra only include the contribution from the discrete/line spectra.
- Setting "-1" instead of providing the energy grid structure disables the option for the given particle type.
set dt
set dt NTRSH [ GTRSH ]
Sets probability threshold for delta-tracking. Input values:
| NTRSH | : probability threshold for neutrons |
| GTRSH | : probability threshold for photons |
Notes:
- Serpent uses delta-tracking by default for both neutrons and photons, but switches to surface-tracking if the probability of sampling virtual collisions (ratio between material total cross section and the majorant) exceeds the given threshold.
- Default probability threshold for both particle types is 0.9, i.e. delta-tracking is used if the ratio between total cross section and majorant is above 0.1.
- "set dt 1" means that delta-tracking is always used and "set dt 0" that it is never used.
- Use of delta-tracking can be enforced or blocked in individual materials using the set forcedt and set blockdt options
- Integral reaction rates are scored using the collision estimator of neutron flux, which has a few adjustable parameters (see set cfe).
- For more information on tracking modes, see the detailed description on delta- and surface-tracking.
set dynccfile
set dynccfile OPT
Option to store precursors and neutrons between time steps in coupled dynamic simulations into a file. Input values:
| OPT | : option to switch on (1/yes) or off (0/no) the store/write dynamic data into a file. Default is on. |
set dynsrc
set dynsrc PATH [ MODE ]
Links previously generated steady state source distributions to be used in a transient simulation with delayed neutron emission.
| PATH | : The path of the previously generated source file (without the .main suffix) |
| MODE | : Precursor tracking mode (0 = mesh based, 1 = point-wise) |
Notes:
- Four source files will be required [PATH].main, [PATH].prec, [PATH].live and [PATH].precpoints
set ecut
set ecut EMINn EMINp
Sets minimum energy cut-off for neutrons and photons. Input values:
| EMINn | : cut-off energy for neutrons (MeV) |
| EMINp | : cut-off energy for photons (MeV) |
Notes:
- The default cut-of energy for photons is 1 keV. Neutron energy cut-off is switched off by default.
- Using energy cut-off for neutrons may lead to non-physical results, since fission and up-scattering may not be accurately modeled.
- Versions 2.1.27 and earlier include only photon energy cut-off, which is now the second input parameter.
set ecutdens
set ecutdens DENS1 EMINp,1 [ DENS2 EMINp,2 ... ]
Sets density-wise minimum energy cut-off for photons. Input values:
| DENSi | : mass density (g/cm3) |
| EMINp,i | : cut-off energy for photons (MeV) |
Notes:
- Mass densities and energy cut-offs must be given in ascending order.
set ecutmat
set ecutmat MAT1 EMINp,1 [ MAT2 EMINp,2 ... ]
Sets material-wise minimum energy cut-off for photons. Input values:
| MATi | : material name |
| EMINp,i | : cut-off energy for photons (MeV) |
set eddi
set eddi OPT
Switches on the calculation of Eddington factors. Input values:
| OPT | : option to switch calculation on (1) or off (0) |
Notes:
- Requires group constant generation to be set on.
set edepdel
set edepdel OPT [ LOCAL_EGD ]
Option to include the energy of delayed components in energy deposition calculations. Input values:
| OPT | : include (1) or exclude (0) the energy of delayed components in energy deposition estimates |
| LOCAL_EGD | : deposit the energy of the delayed fission gammas to fission sites (1) or with the same distribution as the prompt fission gammas (0) (this option is used only in energy deposition mode 3) |
Notes:
- Delayed components include delayed neutrons, delayed fission gammas and delayed betas.
- The energy of delayed neutrons can be excluded using this option only in energy deposition mode 1.
- The energy of the delayed components is deposited at the time of fission so the time dependence of the energy deposition is not accounted for properly in transient simulations.
- Default options are to include delayed components (1) and to deposit the energy of the delayed fission gammas with the same distribution as the prompt fission gammas (0).
set edepkcorr
set edepkcorr OPT
Option to apply correction for energy deposition estimates when simulating non-critical systems with criticality source mode. Input values:
| OPT | : apply (1) or do not apply (0) correction |
Notes:
- The methodology is described in related paper. [4]
set edepmode
set edepmode MODE [ E_CAPT ]
Sets energy deposition mode for energy deposition calculations. Input values:
| MODE | : energy deposition mode (0, 1, 2 or 3) |
| E_CAPT | : additional energy release in capture reactions given in MeVs per fission (used only in energy deposition mode 1) |
Notes:
- The energy deposition modes are described in related paper. [4].
- The choice of energy deposition mode affects also the normalization of the results, if normalization to total power or power density is used.
- Energy deposition modes 1, 2 and 3 require data which is not available in the standard ACE-format cross section files used by Serpent. Separately distributed ACE-files containing additional data are required to use these modes.
- Default option is 0 which corresponds to the methodology used before version 2.1.31.
- KERMA coefficients used in energy deposition modes 2 and 3 are not Doppler-broadened correctly by the built-in preprocessor. See Doppler-broadening preprocessor.
set egrid
set egrid TOL [ EMIN EMAX ]
Sets the unionized energy grid reconstruction parameters. Input values:
| TOL | : fractional reconstruction tolerance |
| EMIN | : minimum energy in the grid (MeV) |
| EMAX | : maximum energy in the grid (MeV) |
Notes:
- The default fractional reconstruction tolerance is 0.0 in transport calculation mode and 5E-5 in burnup calculation mode.
- A higher energy grid reconstruction tolerance means lower memory consumption and possibly higher computation speed but also reduced accuracy of the calculation.
- The default minimum energy is 1E-11 MeV (neutrons) and 1E-03 MeV (photons).
- The default maximum energy is 20.0 MeV (neutrons) and 100.0 MeV (photons).
- See also Section 5.3 of Serpent 1 User Manual.
set ekn
set ekn E
Sets the Klein-Nishina equation to approximate a Compton scattering event. Input values:
| E | : energy cut-off for modelling energy and direction of the scattered photon (MeV) |
Notes:
- The default value of E is set to INFTY (1E+37).
- The Klein-Nishina equation is used above E for calculating both the energy and direction of the scattered photon. Below E, the Doppler brodening method is used if switched on. Otherwise, the incoherent scattering function approximation is in use.
set elcond
set elcond MAT1 COND1 [ MAT2 COND2 ... ]
Sets material-wise conductivity state for electrons in photon transport calculations. Input values:
| MATi | : material name |
| CONDi | : conductivity state (0 = non-conductor, 1 = conductor, 2 = conduction electron dependent). |
Notes:
- The default option is 2 or conduction electron dependent.
- If the material is set as a conductor and no-conduction electrons are found, the material conductivity state is overwritten to non-conductor.
- Option 2, conduction electron dependent, establishes that a single element material is a conductor if conduction electrons are found, otherwise is a non-conductor. A compound is always a non-conductor.
set elgas
set elgas MAT1 GAS1 [ MAT2 GAS2 ... ]
Sets material-wise phase for electrons in photon transport calculations. Input options:
| MATi | : material name |
| GASi | : phase state (0 = condensed or non-gas, 1 = gas). |
Notes:
- The default option is 0/gas and only affects mixtures.
- The gas phase does not affect the mean excitation energy of a single material: if set elmee option is set for a material, the material is considered as non-gas.
set elmee
set elmee MAT1 MEE1 [ MAT2 MEE2 ... ]
Sets material-wise mean excitation energy for electrons in photon transport calculations. Input values:
| MATi | : material name |
| MEEi | : electrons mean excitation energy (MeV) |
Notes:
- The default value is -1.0 interpreted as calculated during runtime for compounds and extracted from data for single element materials.
- The maximum mean excitation energy for electrons is 1 MeV.
set elspn
set elspn EGRID_E
Sets the stopping power energy grid size for electrons/positrons in photon transport calculations. Input values:
| EGRID_E | : energy grid size (default value is 200) |
Notes:
- The thick-target bremsstrahlung model (set ttb option) assumes that the energy array is uniformly distributed in a log-energy scale.
set entr
set entr NX NY NZ [ XMIN XMAX YMIN YMAX ZMIN ZMAX ]
Defines the mesh structure used for calculating fission source entropy. Input values:
| NX | : number of mesh cells in x-direction |
| NY | : number of mesh cells in y-direction |
| NZ | : number of mesh cells in z-direction |
| XMIN | : minimum mesh boundary in x-direction |
| XMAX | : maximum mesh boundary in x-direction |
| YMIN | : minimum mesh boundary in y-direction |
| YMAX | : maximum mesh boundary in y-direction |
| ZMIN | : minimum mesh boundary in z-direction |
| ZMAX | : maximum mesh boundary in z-direction |
Notes:
- Shannon entropy is used to monitor fission source convergence by recording the distribution of source points on mesh.
- The calculation is invoked by setting the generation history record option on (set his).
- The default mesh size is 5 × 5 × 5, extending over the entire geometry.
- Monitoring fission source convergence make sense only in criticality source mode.
- For more information, see detailed description on fission source convergence.
set fininitfile
set fininitfile FILEPATH
Links a file containing an initial fuel behavior solution used as a starting point for a coupled calculation with the FINIX fuel behavior module. Input values:
| FILEPATH | : path to the file |
Notes:
- The file should be a type 6 fuel behavior interface containing a previous solution from a coupled FINIX calculation.
- The axial and radial nodalization as well as the included fuel rods should be the same in the file as in the calculation.
set fissh
set fissh ZAI1 E1 ZAI2 E2 ...
Overrides default fission heating values. Input values:
| ZAIn | : nuclide identifiers (ZAI) |
| En | : energy deposited per fission in MeV |
Notes:
- The energy deposited per fission includes additional energy released in capture reactions when fission neutrons are absorbed.
- By default the energy release per U-235 fission is set to 202.27 MeV, and the values for other actinides scaled based the Q-values found in the cross section libraries.
- See also set U235H.
- See also Section 5.8 of Serpent 1 User Manual.
set fissrate
set fissrate F [ MAT ]
Sets normalization to fission rate.
| F | : number of fission reactions per second (1/s) |
| MAT | : dummy parameter |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set powdens, set flux, set genrate, set absrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set fissye
set fissye OPT
Option to include energy-dependent fission yields. Input values:
| OPT | : include (1/yes) or exclude (0/no) energy-dependent fission yields. Default is 1. |
set flux
set flux F [ MAT ]
Sets normalization to total flux.
| F | : flux |
| MAT | : dummy parameter |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set powdens, set genrate, set fissrate, set absrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set fluxlimtrc
set fluxlimtrc OPT
Option that enables the calculation of flux limited TRC diffusion coefficients. Input values:
| OPT | : option to switch calculation on (1/yes) or off (0/no). Default is off. |
Notes:
- In infinite-spectrum calculations the anisotropic component of the non-TRC nuclides/materials is removed
- S1 and SP1 are non-corrected for non-TRC nuclides/materials and below energy limit for TRC nuclides/materials (see set trc option)
set fmtx
set fmtx 1 MAT1 MAT2 ...
set fmtx 2 UNI1 UNI2 ...
set fmtx 3 LVL
set fmtx 4 XMIN XMAX NX YMIN YMAX NY ZMIN ZMAX NZ
Set the calculation of fission matrixes. Input values:
| MATn | : material list (type 1) |
| UNIn | : universe list (type 2) |
| LVL | : level number (type 3) |
| XMIN | : minimum x-coordinate mesh boundary (type 4) |
| XMAX | : maximum x-coordinate mesh boundary (type 4) |
| NX | : number of x-mesh cells (type 4) |
| YMIN | : minimum y-coordinate mesh boundary (type 4) |
| YMAX | : maximum y-coordinate mesh boundary (type 4) |
| NY | : number of y-mesh cells (type 4) |
| ZMIN | : minimum z-coordinate mesh boundary (type 4) |
| ZMAX | : maximum z-coordinate mesh boundary (type 4) |
| NZ | : number of z-mesh cells (type 4) |
Notes:
- There are four options for defining the regions that compose the matrix: 1 = material, 2 = universe, 3 = level, 4 = Cartesian mesh.
- The output is printed in file [input]_fmtx[bu].m, where "bu" is the burnup step. Output values are given in pairs: matrix element and associated relative error, for total, prompt and delayed (analog) fission matrixes.
set forcedt
set forcedt MAT1 MAT2 ...
Defines the list of materials where delta-tracking is always used. Input values:
| MATn | : material names |
Notes:
- This option is used to override selection of tracking mode based on the probability threshold (see set dt) in individual materials.
- Use of delta-tracking can be blocked in individual materials using set blockdt.
- For more information on tracking modes, see the detailed description on delta- and surface-tracking.
- Note to developers: should have different lists for neutrons and photons?
set fpcut
set fpcut FPCUT
Fission product yield cut-off FPCUT acts on cumulative fission yields, and the given number is the lower limit for the maximum cumulative yield in each mass chain. So "set fpcut 1E-2" means that every mass chain (nuclides with same mass number) with all cumulative yields below 1% are discarded from the calculation.
The cut-off is set to zero by default. Setting the FPCUT cut-off to a higher value (~1E-4 or so) is an effective way to reduce the number of nuclides in the calculation, but at some point it will start affecting the results.
set fsp
set fsp OPT NSKIP
Sets fission source passing between two transport simulations in burnup or coupled calculation. The fission source at the end of one transport calculation is used as the initial source for the next transport calculation.
| OPT | : option to switch fission source passing on (1/yes) or off (0/no) |
| NSKIP | : number of inactive generations on subsequent steps |
Notes:
- Fission source passing is turned off by default.
- Number of inactive generations is taken from set pop card on the first step and from set fsp on all later steps.
set fum
set fum ERG [ BTCH MODE LIM ]
set fum ERG [ BTCH MODE LIM TGT ITER INIT ]
set fum ERG [ BTCH MODE DC LIM TGT ITER INIT ]
Activates fundamental mode calculation for collapsing intermediate multi-group constant data into few-group constants with a critical spectrum.
| ERG | : Intermediate multi-group structure for calculation of the leakage-corrected critical spectrum |
| BTCH | : When set to 2, results are averaged over all criticality cycles |
| MODE | : Critical spectrum calculation type (default 0, i.e. old B1 calculation) |
| DC | : Multi-group diffusion coefficients to be used with FM fundamental mode calculation (default value INF_DIFFCOEF). This value is not read with other modes. |
| LIM | : Convergence criterion of keff in fundamental mode calculation calculated as the absolute value difference of keff between successive iterations (default value 1E-7) |
| TGT | : Target value for fundamental mode keff (default value 1.0, not used with the old B1 calculation mode) |
| ITER | : Maximum number of fundamental mode calculation iterations (default value 25, not used with the old B1 calculation mode) |
| INIT | : First guess for absolute value of critical B2 (default value 1E-6, not used with the old B1 calculation mode) |
The possible values for mode are:
| Mode | Description |
|---|---|
| o, O, 0 | Old B1 calculation (use the same method as before version 2.1.31) |
| b, B, 1 | New B1 calculation |
| p, P, 2 | P1 calculation |
| f, F, 3 | FM calculation |
The possible values for FM mode multi-group diffusion coefficients are:
| Mode | Description |
|---|---|
| 1 | Use out-scattering diffusion coefficients |
| 2 | Use transport corrected diffusion coefficients |
| 3 | Use cumulative migration method diffusion coefficients |
Notes:
- When invoked, Serpent applies a fundamental mode leakage correction on group constants and produces a second set of output parameters.
- Before version 2.1.31, the mode parameter was not read, and the old B1 calculation mode was always used regardless of the parameter given here.
- The group constants are named with B1_ prefix, but they are calculated with fundamental mode calculation type defined with the mode parameter.
- The multi-group structure may be an energy grid defined using the ene card or a name of a pre-defined energy group structure.
- Setting the fundamental mode calculation overrides the default 70-group structure used for macroscopic data calculation in infinite spectrum.
- Averaging the results over all cycles may improve convergence and speed up the calculation, but all information on statistical errors is lost.
- This option does not affect the flux spectrum used during burnup calculations.
- The calculation modes other than the old B1 calculation mode were added in version 2.1.31.
- The descriptions of the other methods than the old B1 method will be presented later in a publication.
- Note that in general the intermediate multi-group structure should have more groups than the few-group structure to get reasonable results for leakage corrected group constants and out-scatter diffusion coefficients.
- Mode 0 (old B1 calculation) might be very slow with a large intermediate multi-group structure (for example with approximately 2000 groups). Modes 1-3 should run much faster.
set gbuf
set gbuf FAC [ BNK ]
Sets the size of photon buffer and event bank. Input values:
| FAC | : factor (> 1) defining the buffer size |
| BNK | : event bank size |
Notes:
- Photon buffer refers to pre-allocated memory block used to store photon particle data. This memory is needed for putting secondary photons in que, etc..
- The buffer factor defines the buffer size relative to simulated batch size.
- The event bank refers to pre-allocated memory block used to store history data on particle events. This bank is used only with certain special options, such as importance detectors and track plotter.
- The default values depend on simulation mode, and there is no need to adjust the values unless the calculation terminates with an error.
- Note to developers: event bank is now the same for both neutrons and photons.
set gct
set gct OPT
Switches on the calculation of group constant statistics test. Input values:
| OPT | : option to switch calculation on (1/yes) or off (0/no). Default is off. |
Notes:
- When this option is set, the batch-wise statistical tests are printed in the file [input]_stat.m.
- Requires group constant generation to be set on.
- Note to developers: statistical tests should be documented
set gcu
set gcu UNI1 [ UNI2 UNI3 ... ]
Sets the universes for group constant generation. Input values:
| UNIn | : universe where group constants are generated or -1 to switch group constant generation off. |
Notes:
- Group constants are by default generated in the root universe (universe 0).
- Group constant generation should be switched off when the results are not needed (this may speed up the calculation).
- See separate description of output parameters.
- Super-imposed group constant generation universes (i.e. not part of the geometry definition) should not be used so that they cover (partly or totally) the same geometry region as some other group constant generation universe (super-imposed or part of the geometry definition), if group constants are generated only on a single geometry level.
set gcut
set gcut GMAX
Sets generation cut-off for neutrons. Input values:
| gmax | : number of simulated generations before cut-off |
Notes:
- The generation cut-off can be used in neutron external source simulations, to limit the length of fission chains.
- Applicable only to neutron external source simulation (invoked using set nps)
- Generation or time cut-off (set tcut) is always needed for neutron external source simulations in super-critical systems.
set genrate
set genrate G [ MAT ]
Sets normalization to fission neutron generation rate.
| G | : number of fission neutrons emitted per second (in neutrons/s) |
| MAT | : material in which the fission neutrons are generated |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- If the material name is omitted, the value corresponds to total fission neutron generation rate in the system.
- The neutron generation rate includes only prompt and delayed neutrons emitted in fission.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set powdens, set flux, set fissrate, set absrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set gpop
set gpop MAX_HIS MAX_POP C
Sets the on-the-fly neutron growing population size algorithm.
| MAX_HIS | : maximum number of histories |
| MAX_POP | : maximum population size |
| C | : parameter to control the population growth |
Notes:
- The on-the-fly neutron growing population size algorithm is used in criticality calculations to accelerate the fission source convergence.
- The growing algorithm methodology is described in related paper. [5]
set gsw
set gsw FILE [ OPT ]
Writes secondary photon source points in coupled neutron-photon transport simulation into a file. Input values:
| FILE | : file name where the source points are written |
| OPT | : option to include secondary photons in transport calculation (1/yes), (0/no) |
Notes:
- Applicable only to coupled neutron-photon transport simulation (invoked using set ngamma).
- Default photon-production (from neutron reactions) transported simulation option is switched off (0/no).
- Only source points from active cycles are included in criticality source simulations.
set his
set his OPT
Sets batch history record on or off. Input values:
| OPT | : option to switch batch history record on (1/yes) or off (0/no) |
Notes:
- When invoked, Serpent collects batch-wise data on keff, fission source entropy etc. and produces a separate output file.
- Setting the history option also invokes the calculation of fission source (Shannon) entropy. The entropy mesh parameters can be adjusted using set entr.
set ifp
set ifp GEN
Sets the number of generations to use for calculating the iterated fission probability. Input values:
| GEN | : Number of generations. Default value 15. |
set imp
set imp TYPE G
Defines energy-dependent importances. Input values:
| TYPE | : type of dependency (e = energy) |
| G | : exponential for adjusting importances (default value: -0.5) |
set imp TYPE r1 I1 [ r2 I2 ... ]
Defines geometry-dependent importances for given region(s). Input values:
| TYPE | : type of (geometrical) dependency (c = cell, m = material, u = universe) |
| rn | : region identifier (cell, material or universe name) |
| In | : importance |
Notes:
- Geometry-dependent importances (only) used for variance reduction
- Only cell (geometry-dependent) type importances supported for now
- Geometry-dependent importances requires using surface-tracking (see set dt)
set impl
set impl ICAPT [ INXN INUBAR ILEAK ]
Sets implicit reaction modes on or off.
| ICAPT | : option to switch implicit capture reactions on (1/yes) or off (0/no) |
| INXN | : option to switch implicit nxn reactions on (1/yes) or off (0/no) |
| INUBAR | : number of fission neutrons to emit in each fission (nonzero = implicit treatment, 0 = analog treatment) |
| ILEAK | : option to switch implicit implicit leakage on (1/yes) or off (0/no) |
Notes:
- Group constant generation requires implicit nxn reactions to be set on.
- If an implicit nubar is given, the weights of the fission neutrons are scaled to conserve the physical number of fission neutrons.
- Implicit leakage requires group constant generation to be set on.
- See separate description of physics options in Serpent for differences to other codes.
set inftrk
set inftrk LOOPn [ ERRn LOOPp ERRp ]
Sets parameters for terminating infinite tracking loops.
| LOOPn | : number of neutron tracking loops interpreted as a geometry error |
| ERRn | : flag to terminate neutron tracking when an infinite loop occurs (0 = no, 1 = yes) |
| LOOPp | : number of photon tracking loops interpreted as a geometry error |
| ERRp | : flag to terminate photon tracking when an infinite loop occurs (0 = no, 1 = yes) |
Notes:
- Serpent checks for tracking loop length to avoid simulation being stuck in an infinite loop. The simulation is terminated by default if no material is found after the particle has been moved forward 1000000 times.
- Long loops can occur by chance in complicated geometries, and this parameter allows continuing the simulation without terminating with error message.
- Even if the problem can be solved by switching the infinite loop error off, it is advised to check the geometry for possible errors.
set inventory
set inventory MAT1 MAT2 ...
set inventory top N PARA
Specifies which nuclides or elements to include in the [input]_dep.m output file. Input values:
| MATn | : Nuclide or element to include |
| N | : Number of nuclides |
| PARAM | : Parameter name |
Notes:
- MAT can be: an isotope, element, or a special entry.
- Isotopes are entered using element symbol and mass number (e.g U-235, Am-242m, etc.) or ZAI (922350, 952421, etc.). In the ZAI format the last digit refers to the isomeric state (0 = ground state, 1 = isomeric state).
- Elements are entered using symbol or numerical (U, 92, etc.). The output includes the sum over all isotopes.
- Special entries include:
- "all" -- All nuclides.
- "accident" -- Nuclides with significant health impact & high migration probability in accident conditions. Source Chernobyl: Assessment of Radiological and Health Impact
- "actinides" -- Actinides (Z>88) for which cross section data are found in JEFF-3.1.1
- "burnupcredit" -- Nuclides commonly considered in burnup credit criticality analyses for PWR fuels [1]
- "burnupindicators" -- Burnup indicators (commonly measured from spent fuel) [2]
- "cosi6" -- Inventory list used by the COSI6 code (excluding lumped fission products)
- "lanthanides" -- Lanthanides (56<Z<72) for which cross section data are found in JEFF-3.1.1
- "longterm" -- Relevant radionuclides in long-term waste analyses [6]
- "minoractinides" -- Minor actinides (actinides - thorium - uranium - plutonium) for which cross section data are found in JEFF-3.1.1
- "fp" -- Fission products
- "dp" -- Actinide decay products
- "ng" -- Noble gases
- The second syntax allows listing the most significant contributors to a given result. The parameters are:
- "mass" -- contribution to mass fraction
- "activity" -- contribution to activity
- "sf" -- contribution to spontaneous fission rate
- "gsrc" -- contribution to gamma emission rate
- "dh" -- contribution to decay heat
- "ingtox" -- contribution to ingestion toxicity
- "inhtox" -- contribution to inhallation toxicity
- for example "top 10 dh" gives the top 10 contributors to decay heat.
- Since most significant contributors may change over time, the output may contain more nuclides than is requested in the input card.
- The calculation of top contributors does not work with the "-rdep" command line option.
set isobra
set isobra ZAI1 MT1 FG1
ZAI2 MT2 FG2
...
Defines constant branching ratios to isomeric states. Input values:
| ZAIn | : nuclide identifiers (ZAI) |
| MTn | : ENDF reaction MT |
| FGn | : fraction of reactions leading to the ground state of the product nuclide |
Notes:
- Serpent uses constant branching ratios by default. This option overrides the default values.
- Energy-dependent data read read from ENDF format files defined by the set bralib overrides the constant ratios.
set iter alb
set iter alb [ CYCLES KEFF FX FY FZ ]
Option that iterates the albedo boundary conditions given a target k-eff for the evaluation. Input values:
| CYCLES | : number of additional inactive cycles to run for the convergence of the iteration (default value: 50) |
| KEFF | : target keff for the iteration (default value: 1.0) |
| FX | : albedo factor in x-direction to normalize the albedo leakage rate (default value: 1.0) |
| FY | : albedo factor in y-direction to normalize the albedo leakage rate (default value: 1.0) |
| FZ | : albedo factor in z-direction to normalize the albedo leakage rate (default value: 1.0) |
Notes:
- The albedo boundary conditions result from the product of the iterated albedo leakage rate by the albedo factor in each direction.
set iter nuc
set iter nuc CYCLES KEFF NZAI ZAI1 ZAI2 ... ZAINZAI [ NMAT MAT1 MAT2 ... MAXNMAT ]
| CYCLES | : number of additional inactive cycles to run for the convergence of the iteration |
| KEFF | : target keff for the iteration |
| NZAI | : number of different nuclides (ZAI) included in the iteration |
| ZAIi | : the ZAI of the nuclide to be included in the iteration (e.g. 50100 for boron 10 ground state) |
| NMAT | : number of different materials included in the iteration (optional parameter) |
| MATi | : the name of the material to be included in the iteration |
Notes:
- If a list of materials is not given, all materials that contain the included nuclides are included in the iteration.
- The initial density of the nuclides to be iterated should be larger than zero.
- The critical density iteration only works for nuclides that have a reactivity effect mainly through neutron absorption.
- Specifically, critical densities of fissile, moderating or reflecting nuclides cannot be reliably iterated using this card.
- The atomic density of the nuclides is updated according to the batching interval set in the set pop option.
- Having a large batching interval means that the atomic density may take a large number of cycles to converge.
set keff
set keff KEFF
Option to scale fission neutron production in external source simulations.
| KEFF | : Fission neutron production scaling factor. Default value is 1. |
Notes:
- The k-effective to use for scaling the fission neutron production. Inverse of KEFF will be used as a multiplicative constant for the nubar, i.e. a value of 2.0 will cut the fission neutron production in half.
- Affects both prompt and delayed neutron production from fissions.
- In versions prior to 2.1.31: does not affect delayed neutron precursor production, which will cause unexpected behaviour in Transient simulations that track delayed neutron precursor concentrations.
set lossrate
set lossrate L [ MAT ]
Sets normalization to total loss rate.
| L | : number of lost neutrons per second (neutrons/s) |
| MAT | : dummy parameter |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- Loss rate includes absorption rate and leakage.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set powdens, set flux, set genrate, set fissrate, set absrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set lost
set lost LIM
Option to treat undefined geometry regions as void. Input values:
| LIM | : maximum number of collisions allowed in undefined regions or -1 if no limit is set. |
Notes:
- This option allows the calculation to proceed even if the geometry routine encounters undefined cells.
- The option is intended, for example, for complex geometries in which the boundaries of adjacent cells may not fully coincide.
- When the option is set, the number of lost particles is printed in variable LOST_PARTICLES in the [input]_res.m output file.
- The option should not be used to get away with errors in incomplete or poorly defined geometries.
set maxsplit
set maxsplit MAX MIN
Sets limiting values for splitting and Russian roulette. Input values:
| MAX | : maximum number of splits |
| MIN | : minimum survival probability in Russian roulette |
Notes:
- Used with weight-windows (see weight window mesh definition).
set mbtch
set mbtch N
Adjusts the batch used with MPI data transfer. Input values:
| N | : batch size in double floats (8 bytes) |
Notes:
- The batch size determines the division of data blocks in MPI data transfer. The value may have some effect on parallel performance.
- The batch size is set to 10000 by default.
set mcvol
set mcvol NP
Runs the Monte Carlo volume-checker routine to set material volumes before running the transport simulation. Input values:
| NP | : number of points sampled in the geometry |
Notes:
- The Monte Carlo based volume calculation routine works by sampling random points in the geometry, and counting the number of hits in every material.
- When invoked, all material volumes are overridden by the results given by the checker routine.
- The volume checker can also be used to produce a separate input file for the volume entries (see detailed description on defining material volumes).
set mdep
set mdep UNI VOL N MAT1 MAT2 ... MATN
ZAI1 MT1
ZAI2 MT2
...
Sets parameters for calculating homogenized microscopic cross sections. Input values:
| UNI | : universe where universe where homogenized microscopic cross sections are generated |
| VOL | : volume of the universe |
| N | : number of materials included in the calculation |
| MAT1 MAT2 ... MATN | : material names |
| ZAIi | : nuclide identifier (ZAI) |
| MTi | : ENDF reaction MT |
Notes:
- When this option is set, Serpent calculates homogenized few-group microscopic cross sections for the listed nuclides and reactions.
- The cross sections are always calculated in the actual spectrum of the problem, never with the critical spectrum.
- The results are printed in a separate output file.
- The results can be included in the group constant output by adding MDEP_XS in the set coefpara list.
- If the number of materials is zero, the calculation is carried over all burnable materials.
- If the list of nuclides and reactions is substituted by "all", Serpent will generate the micro-depletion output including all the nuclides and all reactions involved in the calculation at beginning of the simulation aiming to the extract the constant data (stopping the calculation right after).
- The listed materials must be enclosed inside the homogenized universe.
- The calculation requires the material volumes to be correctly set.
- Transmutation reactions to ground and isomeric states can be calculated by adding 'g' and 'm' after the reaction MT (e.g. 102m is the capture cross section corresponding to daughter nuclide being in isomeric state). Reaction rates are calculated to all states by default.
- Fission reactions corresponding to a specific yield in ENDF can be calculated by adding 1, 2, 3, 4, ... after the reaction MT (e.g. 181 is total fission cross section corresponding to first fission product yield) depending on the fission yield data of the nuclide in the data library. This parameter was different before 2.1.32.
- Sum of all capture reactions can be obtained using MT 101
- Some actinides are missing the total fission channel, and setting the MT to 18 produces sum over MT's 19, 20, 21 and 38 (from version 2.1.29 on).
- The nuclide identifiers are entered as ZAI, not ZA. For example, the ZAI for U-235 is 922350 and the ZAI for Am-242m is 952421.
- Parameter VOL was VR in versions before 2.1.32 (volume ratio of materials included in micro depletion to total homogenized region).
- Multiple set mdep cards can be given for a single homogenized universe.
- The microscopic cross section calculation methodology is described in a related article.[7]
set memfrac
set memfrac FRAC
Defines the fraction of total system memory Serpent can allocate to its use. If the fraction is exceeded, the simulation will abort. This is mainly to avert the use of swap-memory, which can make the system unresponsive. Input values:
| FRAC | : the fraction of system memory that Serpent is allowed to use (between 0 and 1) |
Notes:
- The default fraction is 0.8.
- The fraction can also be set via a SERPENT_MEM_FRAC environmental variable.
- Serpent tries to read the system total memory from the first line of the file /proc/meminfo
set mfpcut
set mfpcut MFPMINp
Sets minimum mean-free-path cut-off for photons. Input values:
| MFPMINp | : cut-off mean-free-path for photons (cm) |
set mfpcutdens
set mfpcutdens DENS1 MFPMINp,1 [ DENS2 MFPMINp,2 ... ]
Sets density-wise minimum mean-free-path cut-off for photons. Input values:
| DENSi | : mass density (g/cm3) |
| MFPMINp,i | : cut-off mean-free-path for photons (cm) |
Notes:
- Mass densities and mean-free-paths cut-offs must be given in ascending order.
set mfpcutmat
set mfpcutmat MAT1 MFPMINp,1 [ MAT2 MFPMINp,2 ... ]
Sets material-wise minimum mean-free-path cut-off for photons. Input values:
| MATi | : material name |
| MFPMINp,i | : cut-off mean-free-path for photons (cm) |
set micro
set micro ERG [ BTCH ]
Defines the intermediate multi-group structure used for group constant generation.
| ERG | : Intermediate multi-group structure used for group constant generation |
| BTCH | : When set to 2, results are averaged over all criticality cycles |
Notes:
- Serpent uses an intermediate multi-group structure to calculate homogenized few-group constants. This input parameter is used to override the default 70-group structure used in the calculation.
- The multi-group structure may be an energy grid defined using the ene card or a name of a pre-defined energy group structure.
- Averaging the results over all cycles may improve convergence and speed up the calculation, but all information on statistical errors is lost.
- Note that in general the intermediate multi-group structure should have more groups than the few-group structure to get reasonable results for leakage corrected group constants and out-scatter diffusion coefficients.
set minxs
See set cfe.
set multilevelgcu
set multilevelgcu OPT
Option that enables the generation of group constants in multiple overlapping universes. Input values:
| OPT | : option to switch calculation on (1/yes) or off (0/no). Default is off. |
set mvol
set mvol MAT1 ZONE1,1 VOL1,1
MAT1 ZONE1,2 VOL1,2
...
MAT2 ZONE2,1 VOL2,1
...
Sets the volumes of material regions. Input values:
| MATm | : name of material m |
| ZONEm,n | : index of zone n in material m |
| VOLm,n | : volume of zone n in material m |
Notes:
- This option is used to define material volumes manually. The input card is also produced when the Monte Carlo based volume checker routine is invoked.
- The zone index is related to automated depletion zone division, invoked by the div card. If no division is used, the index must be set to zero for non-burnable materials. For burnable materials the indexing starts from 1. If no division is used, the corresponding index can be found using the Monte Carlo based volume checker routine.
- Another option to define material volumes is to use the vol entry in the material card.
- For more infomation, see detailed description on the definition of material volumes.
set nbuf
set nbuf FAC [ BNK ]
Sets the size of neutron buffer and event bank. Input values:
| FAC | : factor (> 1) defining the buffer size |
| BNK | : event bank size |
Notes:
- Neutron buffer refers to pre-allocated memory block used to store neutron particle data. This memory is needed for banking fission neutrons for the next generation and putting secondary neutrons in que.
- The buffer factor defines the buffer size relative to simulated batch size.
- The event bank refers to pre-allocated memory block used to store history data on particle events. This bank is used only with certain special options, such as importance detectors and track plotter.
- The default values depend on simulation mode, and there is no need to adjust the values unless the calculation terminates with an error.
- With a large number of neutrons per generation, lowering the factor might be necessary due to excessive memory usage. The default values of the factor allows for large fluctuations of the neutron population due to poor statistics, which is seldom an issue with larger neutron populations.
- Note to developers: event bank is now the same for both neutrons and photons.
set nfg
set nfg ERG
Defines the few-group structure used for group constant generation.
| ERG | : Name of the few-group structure used for group constant generation |
set nfg NE
Defines the few-group structure used for group constant generation. This syntax should not be used anymore.
| NE | : Number of energy groups (1/2/4) corresponding to Serpent 1 default 1-, 2- or 4-group structure. |
set nfg NE ENE-1 ENE-2 ... E1
Defines the few-group structure used for group constant generation. This syntax should not be used anymore.
| NE | : Number of energy groups. Must be at least 2 in this format. |
| EN | : Energy group boundary value between groups N and N+1. Values have to be given in ascending order. |
Notes:
- The few-group structure may be an energy grid defined using the ene card or a name of a pre-defined energy group structure.
- The default is a two-group structure with boundary between fast and thermal group set to 0.625 eV.
- Serpent uses an intermediate multi-group structure in the calculation. The default structure consists of 70 groups, and can be changed using the set micro or set fum options.
- Note that in general the intermediate multi-group structure should have more groups than the few-group structure to get reasonable results for leakage corrected group constants and out-scatter diffusion coefficients.
- The few-group structure must be a sub-set of the intermediate multi-group structure.
- See also: group constant output.
- The few-group structure will also be used for example for assembly discontinuity factors and albedos.
set nfylib
set nfylib LIB1 [ LIB2 LIB3 ... ]
Sets the neutron-induced fission yield library file paths. Input values:
| LIBn | : library file paths |
Notes:
- Fission yield libraries are standard ENDF format files containing neutron-induced fission yield data.
- If the file path contains special characters it is advised to enclose it within quotes.
- A default directory path can be set by defining environment variable SERPENT_DATA. The code looks for fission yield data files in this path if not found at the absolute.
- From version 2.2.0 and on, a default neutron-induced fission yield library directory file can be set by defining environment variable SERPENT_NFYLIB. This file will be used if no other path is given with set nfylib.
set ngamma
set ngamma MODE [ WMIN NMAX ]
Sets the coupled neutron-photon transport simulation on. Input values:
| MODE | : simulation mode (0 = off, 1 = analog, 2 = implicit) |
| WMIN | : weight limit for implicit mode (default value: 0.1) |
| NMAX | : maximum number of emitted photons in implicit mode (default value: 10) |
Notes:
- This input card invokes the production of prompt gammas in neutron reactions. The coupled simulation mode requires that both neutron and photon data libraries are defined.
- In analog mode, the average number of secondary photons produced per collision is defined by the ratio of photon production cross section to material total. Each emitted photon assumes the weight of the incident neutron.
- The implicit mode can be used to produce more photons by allowing variation in their statistical weight. The weight limit defines the minimum allowed weight of emitted photons. This method is close to what is used in MCNP.
- Both calculation modes produce photons in all collisions without any correlation to the sampled reaction mode.
- The MODE option was incorrectly described until Dec. 12, 2017.
set nphys
set nphys FISS [ CAPT SCATT ]
Option to set reaction modes for neutrons on and off. Input values:
| FISS | : option to handle fission (0 = not handled, 1 = handled) |
| CAPT | : option to handle capture (0 = not handled, 1 = handled) |
| SCATT | : option to handle scattering (0 = not handled, 1 = handled) |
Notes:
- All reaction modes are handled by default.
- If fission is switched off, it is handled as capture.
set nps
set nps PP [ BTCH TBI ]
Sets parameters for simulated particle population in external source mode. Input values:
| PP | : total number of particles |
| BTCH | : number of batches |
| TBI | : time binning for dynamic mode |
Notes:
- The total number of particles is divided by the given number of batches to give the number of particles per batch.
- Default number of batches is 200.
- Using the nps card sets the mode to external source simulation. Criticality source simulation for neutrons is invoked using set pop. (The two cards are mutually exclusive).
- If time binning is provided, the simulation is run in the dynamic mode with sequential population control. The bin structure is defined using the tme card.
- Running an external source simulation requires a source, defined by the src card. Source definition also sets the transported particle type.
- Neutron external source simulations are limited to sub-critical systems, unless dynamic mode, time cut-off (set tcut) or generation cut-off (set gcut) is invoked.
- Neutron external source simulations in multiplying systems may require adjusting the neutron buffer (set nbuf).
- Delayed neutron emission is switched off by default in neutron external source simulation. Delayed neutrons can be included with set delnu.
- In transient simulations, where an initial transient source is linked using the set dynsrc option, PP particles are sampled for each time interval.
set opti
set opti MODE
Sets the optimization mode which affects the performance and memory usage. Input values:
| MODE | : optimization mode |
The possible settings for mode are:
| MODE | Description |
|---|---|
| 1 | Minimum optimization and small memory usage. Suitable for very large burnup calculation problems involving tens or hundreds of thousands of depletion zones. |
| 2 | Good performance in burnup calculations involving several thousand depletion zones. Suitable for research reactor applications, but not the best choice for group constant generation. |
| 3 | Similar to mode 4, but lower memory demand. CPU time required by burnup and processing routines increases steeply along with the number of depletion zones, which makes the mode better suited for small burnup calculation problems. |
| 4 | Maximum performance at the cost of memory usage. Suitable for group constant generation and 2D assembly burnup calculations with a limited number of depletion zones. |
Notes:
- Default value for the optimization mode is 4.
- The mode 4 is essentially the same as the methodology in Serpent 1.
- The methodology is described in a paper presented in PHYSOR 2012[8].
set outp
set outp INT
Sets the interval (in cycles) for writing simulation output to files. Input values:
| INT | : number of cycles after which the output-files are updated |
Notes:
- Default value is 50.
- In coupled transient simulations the interval refers to time steps rather than batches. By default, the output is written after every 50th time step.
- Affects files such as [input]_res.m and [input]_det.m as well as mesh plots.
set pbuf
set pbuf FAC
Sets the size of the precursors buffer. Input values:
| FAC | : factor (>1) defining the buffer size |
Notes:
- Precursor buffer refers to pre-allocated memory block used to store precursors data. This memory is needed for banking/retrieving precursors for the next generation/time-interval.
- The buffer factor defines the buffer size relative to simulated batch/cycle size.
set pcc
set pcc MODE [ SSP SSC ]
set pcc 5 PRED CORR [ SSP SSC ]
Sets the time integration method in burnup calculation. Input values:
| MODE | : time integration method |
| PRED | : predictor step integration scheme (0 = constant extrapolation, 1 = linear extrapolation) |
| CORR | : corrector step integration scheme (1 = linear interpolation, 2 = quadratic interpolation) |
| SSP | : number of substeps for predictor steps |
| SSC | : number of substeps for corrector steps |
The possible settings for mode are:
| Mode | Predictor method | Corrector method |
|---|---|---|
| 0, CE | Constant extrapolation | - |
| 1, CELI | Constant extrapolation | Linear interpolation |
| 2, LE | Linear extrapolation | - |
| 3, LELI | Linear extrapolation | Linear interpolation |
| 4, LEQI | Linear extrapolation | Quadratic interpolation |
| 5 | Constant or linear extrapolation | Linear or quadratic interpolation |
| 6, CECE | Constant extrapolation | Constant backwards extrapolation |
Notes:
- MODE 0 is also known as "Euler's method".
- MODE 1 is also known as the "old predictor-corrector method".
- MODE 0 and 1 without substeps corresponds to the methods used in Serpent 1.
- The default numbers of predictor and corrector substeps (SSP and SSC, respectively) are 1 for applicable methods.
- The default time integration scheme corresponds to MODE 1 for applicable methods.
- Number of substeps could not be given for constant predictor or corrector before 2.1.32.
- Decay calculations were always calculated with single substep disregarding this input before 2.1.32.
- The first burnup step extrapolation is always constant and with only single substeps before 2.1.32.
- The custom mode, MODE 5, accepts constant or linear extrapolation for the predictor step and linear or quadratic interpolation for the correct step.
set pdatadir
set pdatadir DIR
Sets the file path for auxiliary photon data. Input values:
| DIR | : file path for directory where the data is located |
Notes:
- Serpent uses auxiliary data files for the modelling of photon interaction physics.
- For more information, see instructions on setting up the data libraries.
set poi
set poi OPT VOL [ XE135M ]
Switches the calculation of poison cross sections on or off. Input values:
| OPT | : option to switch the calculation of poison cross sections on (1/yes) or off (0/no) |
| VOL | : volume of the homogenized zone |
| XE135M | : option to treat 135mXe separate from ground state 135Xe (0 = lumped with 135Xe, 1 = separate treatment) |
Notes:
- Poison cross sections include the fission yields and microscopic and macroscopic absorption cross sections of fission product poisons 135Xe and 149Sm, as well as the fission yields and microscopic absorption cross sections of their precursors. The data is part of the homogenized group constant output.
- The calculation requires setting the decay and fission yield library file paths.
- The calculation requires the material volumes to be correctly set.
- The volume is required for calculating microscopic absorption cross sections matching macroscopic absorption cross sections with poison nuclide densities smeared to the homogenization volume.
- Separate treatment for 135mXe requires cross sections for this isotope.
- Parameter VOL was VR (optional) in versions before 2.1.32 (ratio of fuel volume to the volume of the homogenized zone).
set pop
set pop NPG NGEN NSKIP [ K0 BTCH NEIG ]
Sets parameters for simulated neutron population in criticality source mode. Input values:
| NPG | : number of neutrons per generation |
| NGEN | : number of active generations |
| NSKIP | : number of inactive generations |
| K0 | : initial guess for keff |
| BTCH | : batching interval (default value is 1 generation per batch) |
| NEIG | : number of independent parallel eigenvalue calculations (default is 1) |
Notes:
- The simulation is first run for a number of inactive generations to allow the fission source to converge. This is followed by a number of active generations, during which the results are collected. The statistics are divided in batches, and by default each generation forms its own batch.
- Using the pop card sets the mode to criticality source simulation. External source simulation is invoked using set nps. (The two cards are mutually exclusive).
- Convergence of fission source can be monitored using Shannon entropy (input parameters set his and set entr).
- Initial guess for keff is 1.0 by default. Setting the value manually may get the simulation going if it terminates on the first generation because of poor initial guess. The value does not affect fission source convergence.
- See detailed descriptions on fission source convergence and statistical effects of batching.
- The number of neutrons per generation also affects the memory usage together with set nbuf. With a large value, lowering the buffer size might be necessary for the simulation to be runnable.
set powdens
set powdens PDE [ MAT ]
Sets normalization to power density
| PDE | : power density (in kW/g) |
| MAT | : material in which the given power is produced |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- If the material name is omitted, the value corresponds to average power density produced in the system.
- Power density is given in units of kW/g, not W/g used in several other programs. The typical order of magnitude for this parameter in LWR calculations is 20E-3 ... 50E-3.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set flux, set genrate, set fissrate, set absrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set power
set power P [ MAT ]
Sets normalization to total fission power
| P | : fission power (in W) |
| MAT | : material in which the given power is produced |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- If the material name is omitted, the value corresponds to total fission power produced in the system.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set powdens, set flux, set genrate, set fissrate, set absrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set ppid
set ppid PID
Defines the external code process identifier (PID) number to be used for communication in the POSIX-based coupled calculation communications. Input values:
| PID | : Process identifier (PID) of the (parent) process that Serpent should communicate with. |
Notes:
- Setting up a communication mode will enable the coupled calculation mode.
- PID 222 theoretical maximum for 64-bit system
- The communication options set comfile, set ppid and set pport are mutually exclusive, aka, multiple signalling modes are not allowed.
- For more information see: External coupling
set pport
set pport PORT
Defines the external code port number to be used for communication in the SOCKET-based coupled calculation communications. Input values:
| PORT |
Notes:
- Setting up a communication mode will enable the coupled calculation mode.
- The communication options set comfile, set ppid and set pport are mutually exclusive, aka, multiple signalling modes are not allowed.
- For more information see: External coupling
set ppw
set ppw UNI LAT
Turns on the calculation of pin powers and pin power form factors. Input values:
| UNI | : The universe where the pin power distribution is calculated. |
| LAT | : The lattice where the calculation is performed. |
Notes:
- See separate description of output parameters in the [input]_res.m.
- The form factors can be printed to the group constant output with set coefpara.
- Calculation of pin power form factors also requires calculation of assembly discontinuity factors using set adf.
set precsrcf
set precsrcf FACTOR
Set number of pointwise precursors to hold in memory in transient simulations. A multiplicative factor for the number of neutrons per batch, i.e. setting FACTOR to 10 when running 1000 neutrons per batch will keep 10000 pointwise precursors in memory.
| FACTOR | : The factor to multiply the number of neutrons per batch to obtain the number of pointwise precursors to hold in memory. |
Notes:
- Default value is 10.
- The number of pointwise precursors held in memory is controlled to the requested number at time interval boundaries. The physical number of precursors is conserved.
- Storing a too low of a number of pointwise precursors can lead to undersampling of certain precursor groups or parts of geometry.
set precthresh
set precthresh THRESHOLD
Set the weight threshold for creating and storing a new delayed neutron precursor in transient simulations.
| THRESHOLD | : A weight threshold relative to the incoming neutron weight. |
Notes:
- Default value is 1.
- If the weight of the delayed neutron precursor would be below the threshold, Russian roulette is played to either increase the weight to the threshold or not store the precursor at all.
- Setting a lower threshold stores more precursors with lower weight whereas a higher threshold stores fewer precursors with higher weight.
set printelsp
set printelsp OPT
Prints out the stopping power data for electrons/positions generated by the thick-target bremsstrahlung model. Input values:
| OPT | : Option to output the stopping power data from the bremsstrahlung model off (0/no) or on (1/yes), in file [input]_elsp.m. The default option is off. |
set printm
set printm MODE [ FRAC ]
Print material compositions to [input].bumat[bu] file during burnup calculation, where "bu" is the burnup step.
| MODE | : Set printing on (1/yes) or off (0/no). |
| FRAC | : Optional atomic fraction for printing out decay nuclides (nuclides with no transport cross sections). |
Notes:
- Nuclides without transport data are excluded from the output by default.
- A FRAC value 0.0 will print out all decay nuclides, while 1.0 will not print out any decay nuclides. Value FRAC will print out nuclides whose atomic fraction in the material is greater than or equal to FRAC.
set qparam_dbrc
set qparam_dbrc QPARAM
Sets the Q-parameter (confidence interval) for temperature majorants to be used with Doppler-broadening rejection correction (set dbrc option). Input values:
| QPARAM | : confidence interval |
Notes:
- The parameter should be adjusted when the DBRC majorant cross section is exceeded frequently
- The default value is 2E-5 for the revisited majorant (default method) and reasonable values are within [1E-5, 1E-4]
- The default value is 3.0 for the traditional majorant and reasonable values are within [1.0, 10.0]
set qparam_tms
set qparam_tms QPARAM
Sets the Q-parameter (confidence interval) for temperature majorants to be used with target motion sampling method (mat card). Input values:
| QPARAM | : confidence interval |
Notes:
- The parameter should be adjusted when the TMS majorant cross section is exceeded frequently
- The default value is 2E-5 for the revisited majorant (default method) and reasonable values are within [1E-5, 1E-4]
- The default value is 3.0 for the traditional majorant and reasonable values are within [1.0, 10.0]
set relfactor
set relfactor FAC
Sets the underrelaxation factor for the power relaxation used in coupled multi-physics calculations. Input values:
| FAC | : underrelaxation factor |
Notes:
- Setting the underrelaxation factor to 0, disables power relaxation altogether. The power distribution written to the output-files will be unrelaxed and only based on the most recent iteration.
- Default value is 1.0
set repro
set repro MODE
Sets the reproducibility mode in parallel calculations. Input values:
| MODE | : reproducibility mode |
The possible settings for mode are:
| Mode | Description |
|---|---|
| 0 | No reproducibility |
| 1 | Reproducibility with OpenMP parallelization (default value) |
| 2 | Reproducibility with MPI and hybrid OpenMP / MPI parallelization |
Notes:
- The reproducibility in OpenMP parallelization means that the random number sequences are the same in spite of parallelization. This requires that the histories are calculated always in the same order. This is achieved by sorting the fission banks between batches. With large neutron populations per batches the sorting takes a substantial amount of time which might affect the obtained parallel calculation scalability.
- The reproducibility is often a requirement for debugging the program. MODE 2 should only be used for debugging.
set rfr
set rfr STEP FILE [ NFILE ]
set rfr idx I FILE [ NFILE ]
Reads material compositions from a binary restart file. Input values:
| STEP | : burnup step from which the compositions are obtained |
| I | : burnup step index from which the compositions are obtained |
| FILE | : name of the binary restart file |
| NFILE | : number of restart files |
Notes:
- If it is not specified a burnup or days step explicitly by preceding the depletion step value by bu or days, respectively, then, positive values for step are interpreted as burnup units (MWd/kgU) and negative values as time units (days).
- The continue option sets the restart at the latest calculated depletion step.
- This option can be used together with the set rfw feature for applying changes in the modeled system during burnup calculation.
- The name of the binary restart file is invariant to domain decomposition, set dd. It corresponds with the standard name of the restart file (without the _dd[MPIID] suffix).
- This option reads a single restart file except if the domain decomposition feature is in place (reading the multiple/split restart files generated from the domain decomposition calculation).
- If a non-domain decomposition simulation requires reading the multiple/split restart files from a former one, the number of restart files NFILE should be specified. In any other case, the number of domains/MPI tasks should match between the writing and reading simulations. The material depletion zone divisions between the writing and reading simulations should agree too.
set rfw
set rfw OPT [ FILE ]
Writes material compositions in burnup calculation into a binary restart file. Input values:
| OPT | : option to switch writing on (0 = no, >0 = yes; with, 1= all nuclides, 2 = transport nuclides) |
| FILE | : name of the binary restart file. Default value [input].wrk. |
Notes:
- Restart file writing is switched off by default.
- Version 2.2.0 includes the option to select whether to write all nuclides (default behavior in previous versions) or only the transport nuclides. Before OPT was limited to 0/1.
- This option can be used together with the set rfr feature for applying changes in the modeled system during burnup calculation
- This option when is used with the domain decomposition feature, set dd, generates multiple restart files - as many as domains (MPI tasks) are defined; the files are named adding _dd[mpiid] (domain decomposition identifier) to the standard file name.
set rnddec
set rnddec DEC [ FY DH ]
Option to set decay and fission yield data randomized mode on or off. Input values:
| DEC | : option to switch randomized decay constants on (1/yes) or off (0/no) |
| FY | : option to switch randomized fission yields on (1/yes) or off (0/no) |
| DH | : option to switch randomized decay heat on (1/yes) or off (0/no) |
Notes:
- The default mode for either of the options is off.
set root
set root UNI
Sets the root universe. Input values:
| UNI | : universe name |
Notes:
- Root universe is the universe at the lowest level of the geometry hierarchy, and must always be defined. By default the root is set to "0".
- For more information, see detailed description on the universe-based geometry type in Serpent.
set roulette
set roulette W0 P
Sets parameters for weight cut-off and Russian roulette.
| W0 | : minimum particle weight below which cut-off is applied |
| P | : survival probability for Russian roulette (default value: 0.5) |
Notes:
- Weight cut-off is applied after each collision.
- Can be used together with implicit capture, default value: P = 0.001 (see set impl)
set runtme
set runtme T
Sets the maximum running time for the transport simulation. Input values:
| T | : wall-clock running time (minutes) |
Notes:
- When defined, the transport simulation is terminated after the maximum time is reached.
- Setting the parameter does not override the set pop or set nps option.
set samarium
set samarium OPT [ MAT1 MAT2 ... ]
Sets equilibrium samarium calculation on or off:
| OPT | : option to set equilibrium samarium calculation on (1/yes) or off (0/no) |
| MAT1 MAT2 ... | : optional list of materials for which to set the option (on/off). Default is all fissile materials. Setting equilibrium samarium calculation off for a list of materials sets it on for all other fissile materials. |
Notes:
- The equilibrium concentration is calculated on depletion zone basis. You may want to divide your fuel material into depletion zones
- The equilibrium concentration calculation requires the material volumes to be correctly set.
- The equilibrium concentration calculation requires the fission yield and decay libraries.
- The equilibrium concentration of samarium is updated according to the batching interval set in the set pop card. Having a large batching interval means that the equilibrium concentration may take a large number of cycles to converge.
set savesrc
set savesrc PATH [ PN PP NX NY NZ ]
Sets up the creation of an initial source to be used in a dynamic simulation with delayed neutron emission.
| PATH | : The path of the source file to be created |
| PN | : Fraction of tentative neutrons to save (default 1.0). Only affects criticality source simulations. |
| PP | : Fraction of tentative precursors to save (default 1.0). Only affects criticality source simulations. |
| NX | : Precursor mesh size in x-direction (default 1) |
| NY | : Precursor mesh size in y-direction (default 1) |
| NZ | : Precursor mesh size in z-direction (default 1) |
Notes:
- Four source files will be generated [PATH].main, [PATH].prec, [PATH].live and [PATH].precpoints
- If used in criticality source simulation, the system should be critical and the values will correspond to steady state values.
- If used in a dynamic simulation, the values will correspond to end-of-simulation values
- Before version 2.2.0, if you are getting a warning from function WriteSourceFile "P larger than 1", you should lower the PN value.
- See Transient simulations.
set sca
set sca NI NB MSH
MIN1 MAX1 SZ1
MIN2 MAX2 SZ2
MIN3 MAX3 SZ3
[ SUB1 SUB2 SUB3 LIM ]
set sca NI NB MSH
X0 Y0
P NX NY
MIN3 MAX3 SZ3
[ SUB1 SUB2 SUB3 LIM ]
Invokes a response matrix solver to obtain an improved source guess for criticality source simulations.
| NI | : Number of outer iterations |
| NB | : Number of source batches to collect results |
| MSH | : mesh type (1 = Cartesian, 2 = Cylindrical, 4 = x-type hexagonal, 5 = y-type hexagonal) |
| MINn | : minimum mesh boundary (nth coordinate) |
| MAXn | : maximum mesh boundary (nth coordinate) |
| SZn | : number of mesh cells (nth coordinate) |
| X0, Y0 | : mesh center of hexagonal mesh (currently must be centered at the origin) |
| P | : hexagonal cell pitch |
| NX, NY | : hexagonal mesh size |
| SUBn | : number of sub-mesh cells (nth coordinate) |
| LIM | : convergence criterion (typical value 1E-12) |
Notes:
- The solver is used to accelerate fission source convergence.
- Cartesian and cylindrical mesh are defined by outer mesh boundaries and number of mesh cells.
- The coordinate axes 1, 2 and 3 in Cartesian mesh refer to (x,y,z) and in cylindrical mesh to (r,θ,z), with θ given in degrees.
- The hexagonal mesh is defined by mesh center, cell pitch, number of cells in the radial dimensions (similar to the hexagonal lattice) and axial binning.
- The mesh must be defined slightly larger than the geometry (the mesh boundaries should not coincide with the geometry boundaries).
- Sub-division is used to improve the solution inside mesh cells.
- The methodology is described in a related article.[9]
- The methodology is still under development and, the input syntax may change in future updates.
set seed
set seed RNG [ NBTCH ]
Sets the seed value for the random number sequence. Input values:
| RNG | : seed value used for the random number sequence |
| NBTCH | : starting batch (positive value) or history (negative value) |
Notes:
- By default, Serpent gets the RNG seed from system time. This option overrides the value.
set sfbuf
set sfbuf SIZE
Sets the size of the source file buffer including, e.g., criticality source, secondary photon source, detector source or dynamic source modes. Input value:
| SIZE | : buffer size |
set sfrate
set sfrate S [ MAT ]
Sets normalization to total spontaneous fission rate.
| S | : number of spontaneous fission reactions per second (1/s) |
| MAT | : dummy parameter |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set power, set powdens, set flux, set genrate, set fissrate, set absrate, set lossrate, set srcrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set sfylib
set sfylib LIB1 [ LIB2 LIB3 ... ]
Sets the spontaneous fission yield library file paths. Input values:
| LIBn | : library file paths |
Notes:
- Spontaneous fission yield libraries are standard ENDF format files containing spontaneous fission yield data.
- If the file path contains special characters it is advised to enclose it within quotes
set shbuf
set shbuf [ OPT_BUF OPT_RES2 ]
Option to use a shared or private scoring buffer or results array. Input values:
| OPT_BUF | : use shared (1/yes) or private (0/no) scoring buffer (BUF, default private) |
| OPT_RES2 | : use shared (1/yes) or private (0/no) results array (RES2, default shared) |
Notes:
- Serpent stores scores in a temporary buffer, which in OpenMP parallel mode can be either private (each thread writes in its own buffer) or shared (all threads write in same buffer using atomic operations). Using private buffers increases memory usage to some extent, but it should improve scalability since no barriers need to be set to protect memory.
- Memory consumption of shared RES2 is usually larger than that of shared BUF.
set sie
set sie ITER
Chooses the Stochastic Implicit Euler burnup scheme to be used for the burnup calcualtion. Input values:
| ITER | : tolerance (negative value) or maximum number of iterations (positive value) for each burnup step |
Notes:
- The SIE burnup scheme is mainly intended for burnup calculations that develop instabilities using the default predictor corrector methods.
- The convergence criteria is set either by the tolerance or the maximum number of iterations. If the tolerance is given, ITER < 0, the maximum number of iterations is set to 500. If the maximum number iterations is given, ITER > 0, the tolerance is set to 0.0.
set sourcescale
set sourcescale NB SF1 SF2 ...
Defines a time-interval dependent source scaling factor in dynamic external source calculations (set dynsrc). Input values:
| NB | : number of time intervals |
| SFn | : time-interval-wise source scaling factor |
Notes:
- The number of time-intervals NB should match the definition of the time-bin structure (see tme card)
set spa
set spa CMAP [ FRAC ]
Sets parameters for the source point animation. Input values:
| CMAP | : color map used for plotting the source point animation |
| FRAC | : fraction for the color scheme (minimum/maximum) |
Notes:
- The color maps are: 1 - hot; 2 - cold; 4 - jet; 5 - black and white; 6 - hsv; 7 - spring; 8 - summer; 9 - autumn; 10 - winter; 11 - green-purple; 12 - purple-orange; 13 - blue-red. Many of these correspond to what is used in Matlab.
- The default fraction for the color scheme is 1.0.
set spd
set spd Vn Vp
Overrides the speed of simulated particles. Input values:
| Vn | : speed of neutrons (cm/s) |
| Vp | : speed of photons (cm/s) |
Notes:
- This option is intended for adjusting particle speeds for better visualization in track plot animations.
- Adjusting the speed (obviously) results in incorrect estimates for all time constants.
- Exceeding the speed of light causes a fatal error in debug mode.
set srcrate
set srcrate S
Sets normalization to total source rate.
| S | : source rate (in particles per second) |
Notes:
- Normalization is needed to relate the Monte Carlo reaction rate estimates to a user-given parameter.
- Neutron transport simulations are by default normalized to unit total loss rate.
- Photon transport simulations are by default normalized to unit total source rate.
- For other normalization options, see: set powdens, set power, set flux, set genrate, set fissrate, set absrate, set lossrate, set sfrate.
- See also Section 5.8 of Serpent 1 User Manual.
set stl
set stl EXD NLOOP
Sets options for a STL geometry model
| EXD | : facet uncertainty margin or exclusion distance (default: 1E-05) |
| NLOOP | : maximum number of trials (default: 1000) |
Notes:
- Delta-tracking is enforced by default in STL geometries.
set stlfile
set stlfile OPT
Sets option to read and write STL search mesh into a binary file, [input].smh
| OPT | : option to switch reading/writing on (1/yes) or off (0/no) |
Notes:
- STL mesh search file reading/writing is switched off by default.
set tcut
set tcut Tmax
Sets time cut-off for neutrons and photons. Input values:
| Tmax | : time limit for simulated particle histories (in seconds) |
Notes:
- The time cut-off can be used in both neutron and photon external source simulations, to limit the length of particle histories.
- Time or generation cut-off (set gcut) is always needed for neutron external source simulations in super-critical systems.
- Time cut-off is automatically set in the dynamic external source simulation mode.
- Note to developers: this should take independent values for photons and neutrons
set title
set title NAME
Sets a title for the calculation. Input values:
| NAME | : title used for the calculation |
Notes:
- The title is printed in the run-time output. If the title is not set, the input file name is printed instead.
set tpa
set tpa Tmin Tmax TAIL NF EVB
Sets parameters for track plot animation. Input values:
| Tmin | : starting time of track plot animation (in seconds) |
| Tmax | : end time of track plot animation (in seconds) |
| TAIL | : tail length of plotted particles (in cm) |
| NF | : number of frames |
| EVB | : event bank size |
Notes:
- The track plot animation works with the geometry plotter by creating a number of frames that visualize the motion of particles through the geometry.
- The track plotter is invoked using the "-tracks N" command line option, where N is the number of simulated particle histories (if the set tpa option is not defined, the code plots particle tracks in a geometry plot output).
- The routine produces NF frames [input]_trck[n]_frame[m].png, where [input] is the input file name, [n] is an index corresponding to the geometry plot and [m] is the frame index. The frames can be converted into a gif animation using tools like Imagemagick.
- The plotted particles have a tail to better visualize their movement from one collision to the next. The length of this tail is given in centimeters (similar to geometry dimensions). Neutrons are plotted in magenta, photons are plotted in green.
- Storing the simulated collision points requires additional memory, determined by the event bank size. If the given size is insufficient the calculation is terminated with an error message ("Event bank is empty").
- Producing track plot animations may require adjusting the particle buffers (set nbuf and set gbuf).
- The calculation may require a lot of memory for storing the complete simulated histories in neutron-multiplying systems or when particles are split by variance reduction.
- Motion of thermal and fast neutrons cannot be captured simultaneously because of the several orders of magnitude difference in their speed. Thermal systems are best visualized by enforcing all neutrons to travel at the same speed using the set spd option.
- See also detailed description on track plotter and track plot animation.
set transmurea
set transmurea ZAI1 MT1 ZAI2 MT2 ...
Overrides the automatically generated transmutation paths with a user-provided list of reactions. Input values:
| ZAIn | : nuclide identifier. |
| MTn | : reaction identifier (ENDF reaction MT). |
Notes:
- The transmutation chains in burnup calculation are by default generated automatically. This option overrides the behavior by including only the reactions included on the list.
- The reactions are given as nuclide identifier - reaction identifier pairs, for example, the radiative capture cross section of 238U would be 922380 102.
- The nuclide ID can be replaced with "all", in which case all reactions of the type are included.
set transnorm
set transnorm FACTOR
Multiplies the normalization from a transient source linked with set dynsrc by a factor. Input values:
| FACTOR | : factor to multiply the normalization with. |
Notes:
- The normalization in transient simulations is, by default, same as in the transient source generation calculation.
- See separate description of transient simulations.
set transtime
set transtime FLAG
Choose how time dependent transformations are actually evaluated. Input values:
| FLAG | : Possible values 0/1: |
| With 0, time dependent transformations are evaluated using the beginning time of the time-interval being simulated (geometry static for each time interval). | |
| With 1, time dependent transformations are evaluated using the time when the tracking of the neutron being simulated began (geometry static for each neutron lifetime). |
Notes:
- The default option is 0 (geometry static for each time interval).
- Option 1 generally results in a much smoother transformation.
set trc
set trc MAT FILE Emin [ ZAI1 ZAI2 ... ]
Defines transport correction for the calculation of transport cross section and diffusion coefficient. Input values:
| MAT | : material to which the correction is applied |
| FILE | : file path to correction curve data |
| Emin | : minimum energy above which the correction is applied |
| ZAIn | : nuclide identifiers (ZAI) |
Notes:
- The method calculates transport cross sections by multiplying material total cross sections by a user-defined energy-dependent transport correction ratio before collapsing the energy variable. These transport cross sections are used for calculating diffusion coefficients as
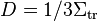 .
. - If the correction ratios are properly defined, the transport cross section is equivalent with the in-scattering approximation.
- The out-scattering approximation of transport cross section is used for materials without defined correction ratios and energies below the given minimum energy.
- The produced homogenized cross sections are written in variables TRC_TRANSPXS and TRC_DIFFCOEF in the [input]_res.m and [input].coe output files.
- See separate description of correction factor file format.
- If nuclide identifiers are given the transport correction is applied only to the total cross sections of these nuclides in the given material. In this case the correction should also be given as the ratio of the sum of transport cross sections of these nuclides and the sum of total cross sections of these nuclides. Otherwise the correction is applied to the total cross section of the material.
- Method can not be used for divided or burnable materials for the time being.
set ttacut
set ttacut TTACUT
Sets cut-off for linear chains method (TTA) based on passage. The cut-off is set to 1E-28 by default. Setting TTACUT cut-off to a higher value serves as a termination criterion where any chain accounting for less than the fraction TTACUT of the total atomic density is ignored.
set ttb
set ttb OPT
Sets the thick-target bremsstrahlung approximation for modelling electrons and positrons on and off. Input values:
| OPT | : option to set thick-target bremsstrahlung approximation off (0/no) or on (1/yes). The default option is on. |
set ttbpm
set ttbpm OPT
Sets a separate bremsstrahlung model for positrons. Input values:
| OPT | : option to set a separate bremsstrahlung model off (0/no) or on (1/yes). The default option is on. |
set ufs
set ufs MODE ORDER NX NY NZ
set ufs MODE ORDER LAT NZ ZMIN ZMAX
set ufs MODE ORDER NX XMIN XMAX NY XMIN XMAX NZ ZMIN ZMAX
Turns on the uniform fission source method. Input values:
| MODE | : reaction rate for the weighted mesh distribution (1 = total (collision), 2 = flux, 3 = fission) |
| ORDER | : exponential factor to adjust the steepness of the distribution |
| NX | : number of x-mesh cells (type 1 and 3) |
| XMIN | : minimum x-coordinate mesh boundary (type 3) |
| XMAX | : maximum x-coordinate mesh boundary (type 3) |
| NY | : number of y-mesh cells (type 1 and 3) |
| YMIN | : minimum y-coordinate mesh boundary (type 3) |
| YMAX | : maximum y-coordinate mesh boundary (type 3) |
| NZ | : number of z-mesh cells (type 1, 2 and 3) |
| ZMIN | : minimum z-coordinate mesh boundary (type 2 and 3) |
| ZMAX | : maximum z-coordinate mesh boundary (type 2 and 3) |
| LAT | : lattice name defining the mesh structure (type 2) |
Notes:
- The UFS method collects the reaction rate distribution during inactive neutron cycles and adjust the number of emitted fission neutrons during the active cycles by a factor for a given mesh cell i:
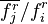 for f the distribution and r the exponential factor.
for f the distribution and r the exponential factor. - The mesh definition can cover the whole geometry (type 1 definition, 3 parameters), be fixed to a lattice (type 2 definition, 4 parameters) or delimited within a region by the number of mesh cells and boundaries in x-, y- and z- directions (type 3 definition, 9 parameters).
- The lattice definition fixes the mesh structure in x- and y- directions. The lattice can be 2D square or hexagonal type defined in the global coordinate system.
- Defining the exponential factor ORDER to 1.0 the method results in a relatively uniform distribution of source points.
- An additional mode, MODE 4, has been set to use the uniform fission method coupled with the built-in response matrix solver. The mesh definition is carried out by the response matrix mesh, see see set sca. (The exponential factor is set to 1.0).
- See Serpent forum topic on UFS method.
set ures
set ures OPT [ NUC1 NUC2 ... ][ DILUCUT ]
Sets unresolved resonance probability table sampling on or off. Input values:
| OPT | : option to switch probability table sampling on (1/yes) or off (0/no) |
| NUCn | : list of nuclides to which the option is applied to, e.g. "92238.09c". Setting unresolved resonance probability table sampling off for a list of nuclides will set it on for the rest of the nuclides. |
| DILUCUT | : infinite dilution cut-off |
Notes:
- Probability table sampling is switched off by default
- The infinite dilution cut-off DILUCUT defines a limit for atomic fractions, and probability table sampling is used only for nuclides with concentration above this limit. The cut-off is set to 1E-09 by default.
- See separate description of physics options in Serpent for differences to other codes.
set usym
set usym UNI AX BC X0 Y0 θ0 θw [ OPT ]
Defines a universe symmetry. Input values:
| UNI | : universe name |
| AX | : symmetry axis (1 = x, 2 = y, 3 = z) |
| BC | : boundary condition (2 = reflective, 3 = periodic) |
| X0 | : x-coordinate of the origin |
| Y0 | : y-coordinate of the origin |
| θ0 | : azimuthal position where the symmetry segment starts (degrees) |
| θw | : width of the segment (degrees) |
| OPT | : option to use actual reflections and translations instead of coordinate transformations |
Notes:
- Universe symmetries can be used to simplify construction of complex geometries.
- Symmetries can also be used to reduce the number of burnable material zones when automated depletion zone division is applied.
- When symmetries are used, it is important to pay attention to the definition of material volumes.
- When using symmetries together with lattices, the lattice positions overlapped by the symmetry should remain empty of fuel pins for example to prevent excess memory usage during depletion calculations.
- Universe symmetries are applied by default using coordinate transformations. This means that the particle position and direction are not affected during tracking. This behavior can be overridden by setting the last option to 1.
- The values of surface current and flux detectors and values produced by for example by set adf cards might not be correct when using actual reflections and transformations.
- Track plots do not work properly with actual reflections and translations.
- For more information, see examples on universe symmetries.
- The supported Serpent 1 syntax-style: set usym UNI SYM [ X0 Y0 ] allows only quadrant symmetries (SYM = 4) in universe 0 (UNI = 0) centred in origin (X0, Y0) = (0,0).
set U235H
set U235H U235_FISSE
Sets the U-235 fission heating value. Input values:
| U235_FISSE | : fission heating value of U-235 (MeV) |
Notes:
- By default the U-235 fission heating value is set to 202.27 MeV and the values for other actinides scaled based the Q-values found in the cross section libraries.
- See also set fissh.
- See also Section 5.8 of Serpent 1 User Manual.
set voidc
set voidc OPT
Option that enables removing void cells to accelerate particle tracking in complex geometries with numerous void. Input values:
| OPT | : option to switch the removal mode on (1/yes) or off (0/no). Default is off. |
set wrnout
set wrnout OPT
Option that enables the generation of a warning-message output file.
| OPT | : option to switch the removal mode on (1/yes) or off (0/no). Default is ON. |
Notes:
- The warning-message output file [input].wrn collects, as a replica, the errors/warnings/notes printed out in the main (screen) output.
set wie
set wie FRAC_ITER
Option that enables the Wielandt’s method to accelerate the convergence of the fission source by setting the initial fraction iteration condition. Input values:
| FRAC_ITER | : initial guess of the probability of banking the neutron (negative value, ]-1, 0[) or the shifted eigenvalue (positive value, [0.5, 3]) |
set wwb
set wwb LO UP [ F ]
Defines the relation between importances and weight-window boundaries. Input values:
| LO | : factor relating the lower weight-window boundary to importance |
| UP | : factor relating the upper weight-window boundary to importance |
| F | : Russian roulette survival probability factor |
Notes:
- Weight window boundaries are inversely proportional to importance. These factors define the coefficients.
- Default values are LO = 0.5, UP = 2.0, F = 1.0.
- See also weight window mesh definition.
set xenon
set xenon OPT [ MAT1 MAT2 ... ]
Sets equilibrium xenon calculation on or off:
| OPT | : option to set equilibrium xenon calculation on or off (0 = off/no, >0 = on/yes; with 1= include only 135Xe, 2 = include 135Xe and 135mXe) |
| MAT1 MAT2 ... | : optional list of materials for which to set the option (on/off). Default is all fissile materials. Setting equilibrium xenon calculation off for a list of materials sets it on for all other fissile materials. |
Notes:
- The equilibrium xenon concentration calculation forces the xenon number density to be in equilibrium with the current flux and xenon absorption level during the transport calculation.
- The equilibrium xenon concentration calculation is meant for example to stabilize the burnup calculations.
- The equilibrium concentration is calculated on depletion zone basis. You may want to divide your fuel material into depletion zones
- The equilibrium concentration calculation requires the material volumes to be correctly set.
- The equilibrium concentration calculation requires the fission yield and decay libraries.
- The equilibrium concentration of xenon is updated according to the batching interval set in the set pop card. Having a large batching interval means that the equilibrium concentration may take a large number of cycles to converge.
set xscalc
set xscalc MODE
Calculation mode for transmutation cross sections. Input values:
| MODE | : Calculation mode (1 = direct tallies, 2 = spectrum-collapse) |
Notes:
- This parameter controls the way transmutation cross sections are calculated in burnup mode. In spectrum-collapse mode these cross sections are calculated after the transport simulation, using a fine-group spectrum collected for each material.
- The spectrum-collapse mode leads to improved performance, but also increased memory footprint per depletion zone.
- The option is automatically set when using optimization modes, and it is not recommended to be defined manually.
- Many of the old example input files set spectrum collapse method on, which overrides the behaviour in lower optimization modes.
- See also set opti.
set xsplot
set xsplot NP Emin Emax
Prints cross section data to [input]_xs0.m file. Input values:
| NP | : Number of energy points to print (minimum value: 10). |
| Emin | : Lower boundary for the energy points. |
| Emax | : Upper boundary for the energy points. |
The cross sections will be printed out at NP logarithmically spaced points between the energy boundaries.
References
- ^ Leppänen, J. "Serpent – a Continuous-energy Monte Carlo Reactor Physics Burnup Calculation Code." User manual, June 18, 2015.
- ^ Leppänen, J. "On the use of delta-tracking and the collision flux estimator in the Serpent 2 Monte Carlo particle transport code." Ann. Nucl. Energy 105 (2017) 161-167.
- ^ Leppänen, J. "Two practical methods for unionized energy grid construction in continuous-energy Monte Carlo neutron transport calculation." Ann. Nucl. Energy 36 (2009) 878-885.
- ^ Tuominen, R., Valtavirta, V. and Leppänen, J. "New energy deposition treatment in the Serpent 2 Monte Carlo transport code." Ann. Nucl. Energy 129 (2019) 224-232.
- ^ Dufek, J. and Tuttelberg, K. "Monte Carlo criticality calculations accelerated by a growing neutron population." Ann. Nucl. Energy 94 (2016) 16-21.
- ^ "The identification of radionuclides relevant to long-term waste management in th United Kingdom", Nirex Report no. N/105 (2004)
- ^ Rintala, A., Valtavirta, V. and Leppänen, J. "Microscopic cross section calculation methodology in the Serpent 2 Monte Carlo code." Ann. Nucl. Energy 164 (2021) 108603
- ^ Leppänen, J. and Isotalo, A. "Burnup calculation methodology in the Serpent 2 Monte Carlo code." In proc. PHYSOR 2012, Knoxville, TN, Apr. 15-20, 2012.
- ^ Leppänen, J. "Acceleration of fission source convergence in the Serpent 2 Monte Carlo code using a response matrix based solution for the initial source distribution." Ann. Nucl. Energy 128 (2019) 63-68